Le appendici auricolari, note in ambito medico anche come orecchio accessorio, trago accessorio o polipo preauricolare, e i seni preauricolari, rappresentano malformazioni congenite benigne che si manifestano in prossimità del padiglione auricolare fin dalla nascita. Sebbene spesso isolate e prive di significative implicazioni funzionali, la loro presenza merita un'attenta valutazione a causa di potenziali associazioni con altre condizioni, comprese sindromi genetiche complesse che possono interessare l'udito e altri organi vitali. Comprendere l'origine, le manifestazioni, le modalità diagnostiche e le opzioni terapeutiche è fondamentale per una gestione ottimale di queste anomalie nel neonato.
Cosa Sono le Appendici Auricolari e i Seni Preauricolari
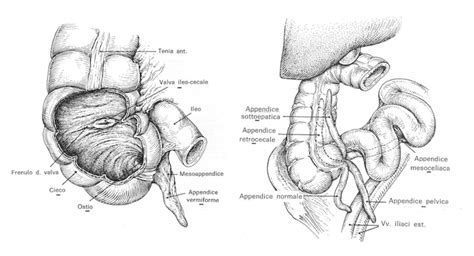
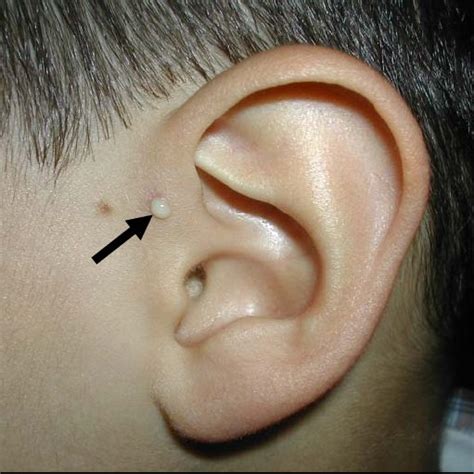
L'appendice auricolare si presenta come una piccola protuberanza di tessuto. Dal punto di vista strutturale, essa è composta da pelle e, molto frequentemente, da un nucleo centrale di cartilagine elastica. La sua posizione tipica è lungo una linea immaginaria che congiunge il trago (la parte rigida davanti al condotto uditivo) all'angolo della bocca (commissura labiale). Clinicamente, l'appendice può presentarsi singolarmente o in gruppi, e può essere unilaterale (interessare un solo orecchio) o, meno frequentemente, bilaterale. Le appendici, escrescenze peduncolate oppure no, singole o multiple, congenite, sono lesioni di norma benigne, localizzate in larga misura al volto, ma con possibile presenza in tutte le sedi corporee.
Il seno preauricolare è una malformazione genetica benigna che consiste in un piccolo foro posto vicino alla cartilagine dell'orecchio, nel punto in cui la cute del viso prosegue con quella che ricopre l'orecchio. Il seno preauricolare può riscontrarsi con la stessa frequenza sia negli uomini che nelle donne e, solitamente, interessa un solo orecchio, anche se, alcune volte, può essere presente bilateralmente.
Queste lesioni, comunemente definite come "pippoli" della cute, hanno una prevalenza di circa 4-5 casi su 1.000 nascituri e sono meritevoli di diversa valutazione, di eventuale trattamento e follow-up nel tempo, a seconda della sede di comparsa o della natura della lesione.
L'Origine Embrionica e le Cause
L'appendice auricolare ha un'origine embriologica ben definita, derivando da un'anomalia durante lo sviluppo dei cosiddetti "tubercoli di His". Questi sei piccoli rigonfiamenti mesenchimali compaiono intorno alla sesta settimana di gestazione. Derivanti dal primo e dal secondo arco branchiale (o faringeo), i tubercoli si fondono tra loro per formare il padiglione auricolare definitivo. Un'anomalia in questo processo porta alla formazione dell'appendice.
Il seno preauricolare è causato da una malformazione genetica benigna che ha origine dall'incompleto assorbimento delle branchie longitudinali (cisti branchiali residue) durante la vita embrionale del feto. Durante lo sviluppo fetale, le creste branchiali, che normalmente si sviluppano in strutture come le orecchie, possono rimanere separate dal resto del sistema e formare il seno preauricolare. Il patologo tedesco Karl Friedrich Van Heusinger documentò per la prima volta questo difetto genetico nel 1864.
Essendo malformazioni genetiche, la predisposizione gioca un ruolo determinante. Sebbene la maggior parte dei casi sia sporadica, ovvero avvenga senza una storia familiare, esistono evidenze di una predisposizione ereditaria. Questa particolare collocazione può rendere ragione di eventuali implicazioni o correlazioni con l'apparato uditivo o profilare quadri malformativi dell'orecchio complessi, dato che i sistemi branchiale, dell'orecchio e dei reni condividono un'origine embrionale comune.
Come funziona l'udito? Fisiologia e anatomia base dell'orecchio
Diagnosi e Valutazione Clinica
La diagnosi di appendice auricolare e seno preauricolare è prevalentemente clinica e viene effettuata dal pediatra o dal neonatologo alla nascita attraverso un semplice esame obiettivo. Si presenta come un nodulo di consistenza variabile, che può essere morbido (se composto solo da pelle e grasso) o rigido (se contiene un asse cartilagineo) per l'appendice, o come un piccolo foro per il seno.
Occorre valutare le caratteristiche del piccolo buco del seno. Ad esempio, se si tratta di un orifizio a fondo cieco, va considerata la possibilità di una infezione locale nel tempo. La presenza di una fistola, invece, desta maggiore attenzione fino alla possibilità di intervento chirurgico, che va eseguito da un professionista con elevata expertise, per la ricchezza di vasi sanguigni arteriosi e venosi e di strutture nervose che caratterizzano la zona.
In passato, si raccomandava routinariamente un'ecografia renale a tutti i bambini con appendici auricolari, a causa della comune origine embrionale di orecchie e reni. Questo perché i bambini con fistole preauricolari presentano un rischio aumentato di sviluppare problemi renali. Pertanto, i medici possono anche eseguire ecografie dei reni.
In alcuni casi, potrebbe essere necessario eseguire ulteriori esami o indagini diagnostiche per confermare la presenza del seno preauricolare e valutarne l'entità, o per escludere altre possibili condizioni. Questi possono includere: ecografia, risonanza magnetica (RM) o tomografia computerizzata (TC). Questi esami di imaging possono essere richiesti per ottenere immagini più dettagliate dell'area circostante al seno preauricolare. In alcuni casi può essere richiesta anche la biopsia per confermare la diagnosi o per escludere altre patologie.
Appendici e Seni Preauricolari: Associazione con Sindromi e Altre Malformazioni
In una piccola percentuale di casi, l'appendice auricolare o il seno preauricolare sono un segno clinico di una sindrome più complessa, il che rende cruciale la valutazione di ulteriori segni associati. Tra le condizioni più rilevanti che possono associarsi a queste malformazioni, troviamo:
Sindrome di Treacher Collins (Disostosi Mandibolo-Facciale): Questa è una malattia genetica rara con malformazioni del volto e problemi di udito. Si stima che un individuo ogni 10-50 mila nasca con la sindrome di Treacher Collins. I bambini presentano dismorfismi facciali caratteristici, con ipoplasia bilaterale e simmetrica delle ossa zigomatiche e del bordo infra-orbitale o della mandibola, che comporta una malocclusione dentale, caratterizzata spesso da un morso mandibolare aperto anteriormente. La sindrome è caratterizzata da anomalie dei padiglioni auricolari (mancanza dell'orecchio, ipoplasia, orecchio a coppa associato a fistole o appendici preauricolari) e anomalie delle palpebre. Altre anomalie meno comuni comprendono la palatoschisi con o senza labioschisi e stenosi/atresia delle narici da un lato solo o da entrambi i lati. È frequente l'ipoacusia trasmissiva (40-50%) causata da alterazioni della catena degli ossicini e sviluppo incompleto dell'orecchio medio. La diagnosi della sindrome di Treacher Collins si pone in base alla presenza di segni clinici maggiori, minori e radiologici. La sindrome è tipicamente causata da mutazioni nel gene TCOF1 (nel 90-95% dei casi), ma possono essere coinvolti anche i geni POLR1B, POLR1BC o POLR1D. Nel 40% dei casi in cui la malattia è a trasmissione autosomica dominante, la mutazione è trasmessa da un genitore (l'alterazione di una sola delle due coppie di geni è sufficiente a provocare la malattia). Nel restante 60% dei casi, la mutazione insorge de novo durante la formazione della cellula uovo o dello spermatozoo o nelle primissime fasi dello sviluppo embrionale.
Sindrome Branchio-Oto-Renale (BOR): Questa sindrome è caratterizzata da anomalie degli archi branchiali (schisi, fistole, cisti), difetti dell'udito (malformazioni dei padiglioni auricolari con fistole preauricolari, sordità di conduzione o neurosensoriale) e anomalie renali (malformazioni delle vie urinarie, ipoplasia o agenesia renale, displasia renale, cisti renali). La prevalenza è stimata a 1 su 40.000. I sintomi più comuni includono difetti delle strutture del collo e del viso, perdita dell'udito, anomalie dell'orecchio esterno e displasia renale. La sindrome è il risultato di disturbi nello sviluppo embrionale delle strutture del collo e dell'orecchio: infatti durante lo sviluppo fetale possono verificarsi alterazioni nelle creste branchiali che portano alla formazione del seno preauricolare e alla manifestazione dei difetti associati alla BOR. La BOR è una condizione genetica ad ereditarietà autosomica dominante, il che significa che una sola copia del gene alterato in ogni cellula è sufficiente a causare il disturbo. I geni coinvolti includono EYA1, SIX1, o SIX5. Le anomalie renali, che si osservano nel 75-85% dei pazienti, possono variare dalla duplicazione del sistema di raccolta alla marcata agenesia del rene, potendo progredire fino all'insufficienza renale a stadio terminale (ESRD) più tardi nella vita.
Sindrome di Beckwith-Wiedemann (BWS): È una rara malattia causata da alterazioni genetiche, principalmente a livello del cromosoma 11, che si manifesta con iperaccrescimento di diversi organi del corpo (lingua grande, visceromegalia ecc.) fin dalla seconda metà della gravidanza e nei primissimi anni di vita. La fistola congenita dell'orecchio, nota come seno preauricolare, è una delle tante espressioni di questa sindrome e può essere collegata a disfunzioni renali.
Sindrome di Goldenhar: In questa condizione, l'appendice preauricolare si inserisce in un quadro di malformazioni monolaterali della mascella, bocca, orecchio e talvolta anche della colonna vertebrale.
In presenza di un'appendice preauricolare o di un seno preauricolare, specialmente se multipli o bilaterali, è consigliabile eseguire un'ecografia renale per escludere anomalie associate, data la comune origine embrionale delle strutture dell'orecchio e dei reni.
Implicazioni per l'Udito: Ipoacusia Associata
La presenza di malformazioni dell'orecchio esterno, come le appendici o i seni preauricolari, può talvolta indicare la possibilità di anomalie più profonde a carico dell'orecchio medio o interno, con conseguente rischio di ipoacusia infantile. L'ipoacusia infantile è un deficit uditivo che può colpire il bambino sin dalla nascita o addirittura nel grembo materno e rappresenta la più frequente disabilità congenita dell'infanzia, manifestandosi in 1-2 nati su mille.
L'indebolimento uditivo causa nel bambino una ridotta percezione dei suoni e dei rumori che lo circondano, causando disorientamento e difficoltà comunicative. Gli effetti sullo sviluppo comunicativo del bambino dipendono dalla sede della lesione, dal grado di perdita uditiva, dall'epoca di insorgenza e dalla causa della perdita uditiva.
I primi segnali di una scarsa capacità uditiva si possono notare già nei primi mesi di vita, con l'assenza di tutte quelle risposte spontanee che si notano nella quotidianità. Il neonato non reagisce a stimoli sensoriali piuttosto forti, come la porta che sbatte o un oggetto pesante che cade a terra, non si gira verso il rumore e rimane indifferente. In presenza di suoni o di melodie non riesce a comprendere da quale parte arrivino.
L'udito è un senso essenziale sin dalla nascita per una corretta acquisizione delle diverse abilità linguistiche e cognitive. Le fasi normali di sviluppo uditivo includono:
- Nascita: Il neonato sente subito i suoni e le voci che lo circondano, anche se risultano ancora molto deboli; in caso di rumori molto forti sussulta e ha una reazione di spavento (piange).
- Dai 2 ai 4 mesi: Comincia a riconoscere il tono di voce alto o basso e la voce dei suoi genitori; capisce di poter emettere da solo dei suoni che lo divertono.
- Dai 5 ai 6 mesi: Il bambino è capace di rispondere a degli stimoli più precisi; cominciano le prime risate, che sono una risposta a percezioni visive o uditive.
- Dagli 8 ai 12 mesi: Riconosce il significato di alcune parole specifiche che rappresentano degli oggetti importanti, come latte e biberon. Se il genitore dice una di queste parole la reazione è immediata.

Fattori di rischio per l'ipoacusia che possono essere correlati alle malformazioni preauricolari:
- Anomalie cranio-facciali: Particolarmente quelle che interessano l'orecchio esterno e il condotto uditivo, come la microtia (orecchio esterno piccolo e deforme) o l'atresia del condotto uditivo esterno (condotto uditivo parzialmente o completamente chiuso). Orecchie ad attaccatura bassa possono essere presenti in varie sindromi genetiche.
- Sindromi associate a sordità: Come la Sindrome di Treacher Collins, la Sindrome di Pendred (ipoacusia bilaterale e gozzo), la Sindrome di Jervell e Lange-Nielsen (sordità neurosensoriale bilaterale profonda congenita e anomalie cardiache) e la Sindrome di Usher (ipoacusia e retinite pigmentosa). La Sindrome di Alport associa ipoacusia e sofferenza renale.
- Cause ereditarie: Il 60% di tutte le ipoacusie riscontrabili nei neonati è dovuto a fattori genetici, che possono essere dominanti (es. gene COCH), recessivi (es. gene connessina 26 GJB2), legati al cromosoma X (es. gene POU3F4) o mitocondriali (es. gene 12rRNA). Molte sindromi associate a malformazioni preauricolari hanno una base genetica.
L'ipoacusia può essere di diversi tipi:
- Ipoacusia trasmissiva: Colpisce l'orecchio esterno o medio. Spesso presente in sindromi come Treacher Collins a causa di alterazioni della catena degli ossicini.
- Ipoacusia neurosensoriale: Spesso congenita, di diversa entità ma permanente, dovuta a malfunzionamenti nel sistema nervoso o dell'orecchio interno.
- Ipoacusia percettiva: Problema che deriva da malfunzionamenti nel sistema nervoso, compromettendo la trasmissione dei segnali nervosi.
- Ipoacusia mista: Combinazione di trasmissiva e neurosensoriale.
Diagnosi e Screening dell'Ipoacusia Infantile (quando associata a malformazioni preauricolari)
Il riconoscimento dei primi segni di un'ipoacusia nei bambini è compito principalmente dei genitori. Per i neonati, sono raccomandati gli esami di screening uditivo prima dei tre mesi di vita. La diagnosi orientativa di alcune forme genetiche potrebbe essere fatta già durante la gravidanza attraverso la villocentesi, una tecnica invasiva che preleva frammenti di placenta per diagnosi prenatale. La positività di questo test consente un intervento precoce subito dopo la nascita.
In caso di sospetto difetto genetico, è possibile eseguire test genetici. La risonanza magnetica per immagini (RMN) può essere eseguita in quasi tutti i bambini per identificare la causa dell'ipoacusia e formulare una prognosi, mostrando ad esempio coclea ipoplasica e canali semicircolari assenti o ipoplasia in alcuni casi sindromici.
Per la diagnosi di ipoacusia nei bambini più grandi esistono diverse tecniche:
- Colloquio con i genitori (anamnestico): Assume un valore diagnostico importante per sapere se il bambino risponde agli stimoli sonori ambientali, quali sono le condizioni del linguaggio, se si comporta come i bambini della sua età, se esiste una consanguineità o la presenza di eventuali parenti sordi.
- Visita con otomicroscopia: Esame diretto dell'orecchio.
- Testare la risposta ai suoni: Nei bambini fra i 6 mesi e i 2 anni d'età.
- Timpanometria: Esamina la risposta del timpano e la funzionalità dell'orecchio medio; può rilevare la presenza di secrezioni nell'orecchio medio.
- Audiometria infantile:
- Tecniche soggettive: Prevedono la collaborazione del bambino. Nei bambini più piccoli (3 mesi-3 anni) si sfruttano reazioni incondizionate o condizionate al suono. Dopo i 3 anni, si può ottenere un valore preciso della soglia delle due orecchie separatamente con strumenti che utilizzano cuffie. Dai 4 ai 5 anni è possibile effettuare l'audiometria convenzionale basata sulle risposte soggettive fornite senza alcun condizionamento.
- Tecniche oggettive: Non richiedono la collaborazione del paziente, come l'impedenzometria e i Potenziali Evocati Uditivi del Tronco Encefalico (ABR).
Le linee guida internazionali (Joint Committee on Infant Hearing, USA, 2007) affermano la necessità di fare diagnosi entro i tre mesi di età e procedere con il trattamento protesico-riabilitativo entro i sei mesi, raggiungendo una protesizzazione ottimale a dodici mesi. Nei casi in cui non si tragga beneficio dalle protesi acustiche tradizionali, si ricorre all'impianto cocleare non prima dei diciotto mesi di vita. La diagnosi presenta spesso molte difficoltà in rapporto all'entità del danno, all'età del bambino e alla possibile presenza di turbe neuropsicologiche associate, e non può mai essere posta con certezza dopo un solo esame audiometrico, ma deve sempre essere fatta dopo una serie di esami e di ripetute indagini.
Come funziona l'udito? Fisiologia e anatomia base dell'orecchio
Trattamento delle Appendici e Seni Preauricolari
Il trattamento dell'appendice auricolare e del seno preauricolare è esclusivamente chirurgico e viene intrapreso principalmente per ragioni estetiche o per prevenire irritazioni croniche, specialmente nel caso dei seni. Generalmente il seno preauricolare non è considerato pericoloso e non richiede trattamento a meno che non causi sintomi o complicazioni.
1. Escissione Chirurgica Classica per Appendici Auricolari:È la procedura d'elezione. Viene eseguita da un chirurgo pediatrico, un chirurgo plastico o un otorinolaringoiatra. L'intervento consiste nella rimozione completa dell'escrescenza cutanea e, soprattutto, della base cartilaginea sottostante. Le tempistiche prevedono che l'intervento sia solitamente programmato dopo i 6-12 mesi di vita. Questo ritardo permette di eseguire l'operazione in anestesia generale con una maggiore sicurezza per il bambino. Studi avrebbero dimostrato che la rimozione immediata, nei primi giorni di vita, non si associa a eventi avversi o a cicatrici nei tre mesi successivi al follow up, ma l'approccio standard rimane quello di attendere.
2. Rimozione Chirurgica per Seni Preauricolari:Le fistole del seno preauricolare vengono rimosse chirurgicamente e l'intervento si svolge di norma in anestesia locale (o generale nei bambini piccoli). Si procede con una incisione cutanea davanti al padiglione auricolare per asportare la fistola. La fistola deve essere asportata con una piccola losanga di cute intorno al suo orifizio. Nella sede di intervento si può lasciare per due o tre giorni un piccolo drenaggio (un tubicino in silicone) per far drenare la secrezione siero-ematica ed evitare che si formi un ematoma (ossia una raccolta di sangue tra i tessuti). L'operazione per rimuovere il seno preauricolare, chiamata "escissione del seno preauricolare", non è dolorosa perché è eseguita in anestesia locale. Il tempo di intervento varia a seconda della dimensione e della complessità del seno preauricolare.
Rischi e Complicazioni:Un seno preauricolare infetto, se non viene adeguatamente trattato, può causare problematiche di varia natura. In alcuni casi, la malformazione può infettarsi o si può formare un ascesso al suo interno. Questo può causare dolore, gonfiore e arrossamento nella zona circostante. In caso di infezioni persistenti è opportuno il consulto da parte di un medico chirurgo specializzato in otorinolaringoiatria o del maxillo facciale. Se si verifica un'infezione, può essere necessario consultare un medico per valutare la situazione e prescrivere antibiotici o drenare l'ascesso. Spesso è necessario intervenire chirurgicamente perché il seno preauricolare è infetto. L'infezione nel seno preauricolare può diffondersi alle strutture circostanti (l'orecchio esterno o il condotto uditivo esterno) causando problemi all'orecchio, come otite esterna.Come tutte le operazioni chirurgiche, l'operazione può comportare dei rischi che sono ben controllati con l'utilizzo di antinfiammatori e antibiotici. I rischi includono cicatrizzazione dolorosa o esuberante della cute (cheloide) e la possibile ricomparsa di una tumefazione o dell'orifizio fistoloso.
Post-operatorio e Recupero:Il post-operatorio è generalmente semplice: si applica una piccola medicazione per pochi giorni e i punti di sutura sono spesso riassorbibili, eliminando la necessità di rimuoverli manualmente. Dopo l'intervento chirurgico, è normale avvertire fastidio nell'area sottoposta a chirurgia. Tuttavia, il dolore solitamente può essere controllato con l'assunzione di analgesici prescritti dal medico. Le tempistiche di guarigione variano da persona a persona, in base al tipo di intervento; in genere si guarisce in due settimane.
Legatura con Filo (Sconsigliata):In passato, alcune appendici molto sottili venivano rimosse legando un filo di seta alla base per interrompere l'afflusso di sangue (necrosi ischemica). Questa metodica non è più raccomandata a causa del rischio di infezioni e cicatrici esteticamente meno favorevoli.
Prognosi e Prevenzione
La prognosi per i bambini nati con un'appendice auricolare isolata è eccellente. Una volta rimossa chirurgicamente, la lesione non si ripresenta e non lascia conseguenze funzionali. Dal punto di vista estetico, i risultati della chirurgia plastica sono ottimi, con un'altissima soddisfazione dei genitori e, in futuro, del paziente stesso. Per i seni preauricolari, se non trattati, la prognosi è buona purché non si verifichino infezioni. È estremamente raro che un paziente possa perdere l'udito a causa di un seno preauricolare infetto non trattato.Nei rari casi in cui l'appendice o il seno facciano parte di una sindrome, il decorso dipenderà dalla gravità delle altre anomalie associate (ad esempio, problemi cardiaci, renali o uditivi), e il follow-up dovrà essere mirato alla gestione di queste condizioni multifattoriali.
Trattandosi di malformazioni congenite legate allo sviluppo embrionale precoce, non esistono misure di prevenzione specifiche per evitare la formazione di un'appendice auricolare o di un seno preauricolare, in quanto si formano nel processo di embriogenesi. Tuttavia, nel caso di peggioramenti dei sintomi, è sempre bene rivolgersi a un medico.
Altre Malformazioni Congenite dell'Orecchio Esterno
Oltre alle appendici e ai seni preauricolari, i difetti congeniti dell'orecchio comprendono altre malformazioni fisiche già presenti nel periodo prenatale, il cui termine "congenito" significa "presente dalla nascita". Queste anomalie possono variare in gravità e avere diverse implicazioni per la funzione uditiva.
- Microtia: Si riferisce a un orecchio esterno (padiglione) piccolo e deforme o scarsamente sviluppato. L'immagine mostra un orecchio esterno piccolo e deforme (microtia), un difetto congenito.
- Atresia del condotto uditivo esterno: Questa condizione implica un condotto uditivo parzialmente o completamente chiuso. Microtia e atresia del condotto uditivo esterno sono spesso concomitanti e di solito vengono identificate al momento del parto o subito dopo. Nei casi gravi, può mancare l'intero condotto uditivo.
- Orecchie ad attaccatura bassa: Sono considerate ad attaccatura bassa le orecchie la cui parte superiore è situata sotto l'angolo esterno dell'occhio. Possono essere presenti in varie sindromi genetiche e i bambini spesso lamentano ritardi dello sviluppo. L'immagine mostra un bambino con orecchie ad attaccatura bassa e altre caratteristiche facciali tipiche di un disturbo genetico.
Questi difetti possono essere segni di altri problemi, pertanto i medici spesso sottopongono il bambino a esami per la perdita dell'udito e per altri difetti congeniti. Inoltre, eseguiranno esami dell'udito per verificare se l'udito è interessato ed esami di diagnostica per immagini del cranio per valutare eventuali problemi ossei.Un bambino con difetti congeniti dell'orecchio può essere valutato da un genetista, un medico specializzato nella scienza dei geni e del modo in cui certe qualità o tratti vengono trasmessi dai genitori alla prole. Possono essere effettuati test genetici di un campione di sangue del neonato per cercare anomalie cromosomiche e genetiche. Questi esami possono aiutare i medici a stabilire se la causa è una specifica malattia genetica e ad escluderne altre.
Il trattamento dei difetti auricolari, inclusi microtia e atresia, può comportare chirurgia ricostruttiva per creare un orecchio esterno di aspetto normale e un condotto uditivo esterno. In molti casi, l'utilizzo di un apparecchio acustico può essere necessario per compensare l'ipoacusia associata.
tags: #appendici #auricolari #neonato