L’analisi citogenetica, nota anche come mappa cromosomica o cariotipo, rappresenta lo studio fondamentale dei cromosomi all'interno delle cellule. Questi ultimi racchiudono i geni, le unità fondamentali costituite da DNA, che portano con sé tutte le informazioni indispensabili per la “costruzione” di un individuo e per il corretto funzionamento dell’intero organismo. Ogni cellula umana, nella sua normalità, ospita 46 cromosomi, frutto della fusione di 23 cromosomi paterni, trasmessi tramite lo spermatozoo, e 23 cromosomi materni, apportati dalla cellula uovo. Gli spermatozoi e le cellule uovo, definiti cellule germinali, sono uniche nel loro genere poiché contengono solo 23 cromosomi. La determinazione del sesso biologico avviene attraverso i cromosomi sessuali: se lo spermatozoo porta il cromosoma X, si svilupperà un individuo di sesso femminile; se porta il cromosoma Y, sarà di sesso maschile. Pertanto, il cariotipo di una femmina normalmente costituito è 46, XX, mentre quello di un maschio è 46, XY.
Scopo e Metodologia dello Studio Citogenetico
Lo studio citogenetico riveste un’importanza cruciale in quanto permette di accertare il numero esatto e la struttura dei cromosomi presenti in un individuo. Le cellule germinali vengono prodotte a partire da cellule somatiche che contengono 46 cromosomi, attraverso un processo complesso di divisione cellulare noto come meiosi. Durante questo processo, possono verificarsi errori nella distribuzione dei cromosomi tra le cellule figlie, un fenomeno denominato non disgiunzione.
L’età materna si configura come uno dei fattori primari che incrementano la probabilità di incorrere in tale non disgiunzione. È inoltre l’unico fattore la cui incidenza è stata scientificamente dimostrata come realmente significativa nella Sindrome di Down, o trisomia 21, condizione caratterizzata dalla presenza di tre copie del cromosoma 21 anziché le due tipiche, portando il numero totale di cromosomi a 47. L’influenza dell’età materna è tangibile: il rischio di avere un figlio affetto da Sindrome di Down per una donna di 25 anni è dello 0,8%, un dato che sale al 2,7% per una donna di 35 anni e raggiunge il 9,2% a 40 anni. Complessivamente, la frequenza delle anomalie cromosomiche tra i neonati si aggira intorno allo 0,5%. Queste anomalie sono alla base di circa un centinaio di differenti condizioni cliniche, tra le quali spiccano per notorietà la Sindrome di Down (trisomia 21), la Sindrome di Edwards (trisomia 18), la Sindrome di Patau (trisomia 13), la Sindrome di Klinefelter (presenza di un cromosoma X in eccesso in soggetti di sesso maschile) e la Sindrome di Turner (assenza di un cromosoma X in soggetti di sesso femminile).
Indicazioni per lo Studio Citogenetico: Prenatale e Postnatale
Lo studio citogenetico trova applicazione in diverse fasi della vita, sia prima che dopo la nascita.
Citogenetica Prenatale
Questa analisi viene eseguita in quelle gravidanze in cui sussiste un rischio aumentato di anomalie cromosomiche nel feto. Le indicazioni includono:
- Età materna pari o superiore ai 35 anni compiuti prima della data prevista del parto.
- Presenza di un figlio affetto da errore nel numero dei cromosomi.
- Genitori portatori di riarrangiamenti cromosomici strutturali che, sebbene non manifestino segni clinici, potrebbero trasmettere alterazioni.
- Genitori con errori nel numero dei cromosomi sessuali (es. 47,XXX; 47,XXY).
- Anomalie fetali evidenziate durante esami ecografici.
- Indicazioni derivanti da specifici test biochimici (come il tri-test).
- Storia di aborti spontanei ripetuti.
Le procedure più comuni per ottenere il materiale genetico fetale sono la villocentesi, eseguibile nel primo trimestre di gravidanza (tra la 9ª e la 12ª settimana di gestazione), e l’amniocentesi, effettuabile nel secondo trimestre (tra la 15ª e la 18ª settimana di gestazione).
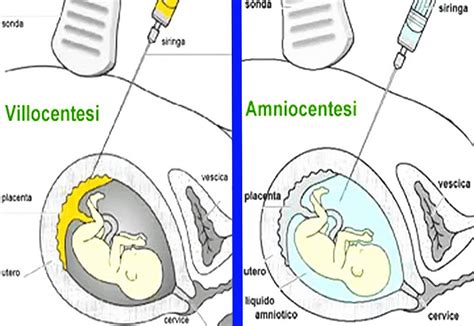
Citogenetica Postnatale
Le indicazioni per l’analisi citogenetica postnatale sono altrettanto variegate e comprendono:
- Soggetti con sospetta sindrome cromosomica, come un neonato che presenta tratti caratteristici della Sindrome di Down, tra cui un aspetto del viso peculiare, anomalie cardiache o la presenza di una singola piega palmare trasversale.
- Genitori e familiari di individui affetti da anomalie cromosomiche.
- Individui con ritardo mentale e/o difetti congeniti non altrimenti spiegati.
- Bambini con ritardo della crescita staturo-ponderale.
- Neonati nati morti, o genitori di soggetti malformati o con sospette sindromi cromosomiche deceduti senza una diagnosi definitiva.
- Coppie con anamnesi di aborti spontanei ripetuti.
- Infertilità maschile di origine non chiara.
- Femmine con amenorrea primaria (mancanza del primo ciclo mestruale) o secondaria (interruzione del ciclo mestruale).
- Situazioni che richiedono fecondazione assistita.
- Sospette sindromi mendeliane, sindromi da geni contigui o sindromi da instabilità cromosomica.
Trisomia 16: Specificità Cliniche e Implicazioni
Il cromosoma 16, come tutti gli altri, viene normalmente ereditato in due copie, una da ciascun genitore. La trisomia 16 completa, ovvero la presenza di tre copie dell’intero cromosoma 16, è una condizione generalmente incompatibile con la vita. Nella stragrande maggioranza dei casi, essa si manifesta con un aborto spontaneo che avviene durante il primo trimestre di gravidanza.
Tuttavia, esiste una rara condizione nota come mosaicismo della trisomia 16. In questo scenario, solo una parte delle cellule dell’organismo presenta la copia extra del cromosoma 16, mentre altre cellule hanno un corredo cromosomico normale. Il mosaicismo della trisomia 16 è una forma di malattia cromosomica che può essere compatibile con la vita, permettendo a un bambino di nascere vivo.
Durante il monitoraggio prenatale, l'analisi dei livelli di trisomia nei tessuti feto-placentari può fornire indicazioni preziose sui potenziali esiti di gravidanze affette da mosaicismo della trisomia 16. Studi condotti su casi di diagnosi prenatale hanno evidenziato che circa il 66% dei nati vivi in queste circostanze presentava un’età gestazionale media di 35,7 settimane. Tuttavia, circa il 45% di questi nati mostrava malformazioni.
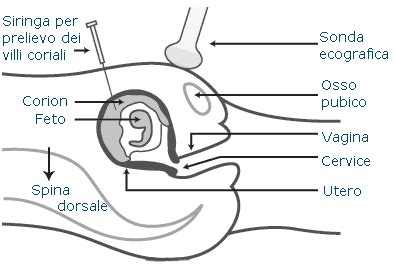
Per la rilevazione della trisomia 16 durante la gestazione, le donne possono sottoporsi a procedure invasive come la villocentesi o l’amniocentesi. Con l'evoluzione delle tecniche non invasive, oggi è possibile utilizzare screening prenatali basati sul sequenziamento di nuova generazione, che analizzano il DNA fetale libero circolante nel sangue materno per rilevare aneuploidie cromosomiche, inclusa la trisomia 16, con elevata sensibilità e specificità.
Duplicazioni del Cromosoma 16 e Anomalie Parziali
Le duplicazioni del cromosoma 16 rappresentano una categoria di anomalie citogenetiche distinte dalla trisomia completa o del mosaicismo. Queste sono caratterizzate dalla presenza di una copia extra di un segmento specifico del materiale genetico sul sedicesimo cromosoma umano. In condizioni fisiologiche, ogni individuo possiede due copie di ciascun cromosoma. Nel caso di una duplicazione parziale, una porzione del cromosoma 16 è presente in tre copie invece delle consuete due.
Il cromosoma 16 è particolarmente predisposto a riarrangiamenti strutturali, inclusa la duplicazione, a causa della presenza di numerose sequenze ripetute di DNA, definite duplicazioni segmentali. Queste sequenze possono favorire l'insorgenza di errori durante i processi di divisione cellulare.
Le conseguenze cliniche derivanti da queste duplicazioni sono estremamente variabili e dipendono in larga misura dalla dimensione del segmento duplicato e dai geni specifici che esso contiene. Dal punto di vista biologico, la presenza di geni in eccesso altera il "dosaggio genico", portando a una sovraespressione di determinate proteine. Queste proteine in sovrannumero possono interferire con i normali processi di sviluppo embrionale, con impatti particolarmente rilevanti sul sistema nervoso centrale, sul sistema cardiovascolare e sull’apparato scheletrico.
ALTERAZIONI CROMOSOMICHE
La causa primaria di queste duplicazioni è solitamente un errore casuale che si verifica durante la formazione delle cellule riproduttive (ovociti o spermatozoi) o nelle primissime fasi dello sviluppo embrionale. Il meccanismo biologico più comunemente implicato è la ricombinazione omologa non allelica (NAHR). Nella maggior parte dei casi, queste anomalie si manifestano de novo, cioè non sono ereditate dai genitori, comportando un rischio di ricorrenza per gravidanze successive molto basso, generalmente inferiore all'1%. Tuttavia, in una percentuale non trascurabile di casi, la duplicazione può essere trasmessa da un genitore portatore della stessa alterazione. Ad oggi, non sono stati identificati specifici fattori di rischio ambientali, comportamentali o legati allo stile di vita che possano indurre in modo specifico le duplicazioni cromosomiche.
Il quadro clinico associato alle duplicazioni del cromosoma 16 è notevolmente eterogeneo. Alcuni individui possono condurre una vita sostanzialmente normale, ignari della loro condizione genetica, mentre altri possono presentare sfide significative sin dalla nascita. La maggior parte dei pazienti manifesta un ritardo dello sviluppo psicomotorio, con una maggiore lentezza nel raggiungimento delle tappe fondamentali quali stare seduti, camminare o parlare. La disabilità intellettiva può variare da lieve a moderata. Sul piano comportamentale, è relativamente comune riscontrare tratti riconducibili al disturbo dello spettro autistico, caratterizzati da difficoltà nella comunicazione sociale e da comportamenti ripetitivi.
Sebbene non esista una "faccia tipica" associata a queste duplicazioni, possono osservarsi lievi dismorfismi facciali, come orecchie a basso impianto, fronte prominente o ipertelorismo (occhi distanziati). La diagnosi di duplicazioni del cromosoma 16 non può basarsi esclusivamente sui segni clinici, poiché i sintomi spesso si sovrappongono a quelli di numerose altre condizioni genetiche.
Diagnosi e Tecnologie Avanzate
La diagnosi definitiva delle duplicazioni del cromosoma 16 si avvale di tecnologie di analisi genetica avanzate:
- Chromosomal Microarray Analysis (CMA): Attualmente considerato l'esame d'elezione (gold standard), questa tecnica, nota anche come array-CGH, è capace di identificare guadagni (duplicazioni) o perdite (delezioni) di materiale genetico anche di dimensioni molto ridotte (microduplicazioni), che sfuggirebbero all'analisi di un cariotipo tradizionale.
- Cariotipo Standard: Questa metodologia è in grado di identificare solo duplicazioni di grandi dimensioni (macroduplicazioni) che sono visibili al microscopio.
La diagnosi può essere effettuata in epoca prenatale tramite villocentesi o amniocentesi, spesso richieste a seguito di riscontri ecografici anomali (come difetti cardiaci o ritardo di crescita intrauterino) o in seguito a screening genetici avanzati.
Il test Prenatal SAFE®, ad esempio, prevede la possibilità opzionale di un approfondimento di secondo livello per identificare la presenza di 6 tra le più comuni sindromi da microdelezione. Queste sono anomalie cromosomiche caratterizzate dalla perdita di un tratto cromosomico di dimensioni ridotte e, di conseguenza, dei geni localizzati su quel frammento.
Trisomia 16 a Mosaico e Diagnosi Prenatale
La trisomia 16 a mosaico, come accennato, è una condizione in cui solo alcune cellule possiedono una copia extra del cromosoma 16. L'analisi dei villi coriali tramite villocentesi o del liquido amniotico tramite amniocentesi può rivelare questa condizione. In uno studio su casi di diagnosi prenatale, è stato osservato che in gravidanze con mosaicismo per la trisomia 16, il 66% risultava in nati vivi, con un’età gestazionale media di 35,7 settimane. Tuttavia, una percentuale significativa (circa il 45%) presentava malformazioni. I livelli di trisomia rilevati nei tessuti feto-placentari possono fungere da predittori dei risultati in queste gravidanze.
Il test Prenatal SAFE Plus non solo valuta le aneuploidie dei cromosomi 21, 18, 13 e dei cromosomi sessuali, ma include anche la trisomia dei cromosomi 9 e 16 (come opzione). Permette inoltre di identificare alterazioni cromosomiche strutturali submicroscopiche, come alcune comuni sindromi da microdelezione. Il test Prenatal SAFE Karyo offre un’analisi ancora più estesa, rilevando aneuploidie e alterazioni cromosomiche strutturali fetali a carico di ogni cromosoma, con risultati paragonabili a quelli ottenuti con il cariotipo fetale tramite tecniche invasive. Questi test sono integrati con RhSafe, un esame non invasivo che determina il Fattore Rh(D) fetale da un campione di sangue materno.
Tutti questi test possono essere eseguiti a partire da un'età gestazionale di almeno 10 settimane. Durante la gravidanza, frammenti di DNA fetale circolano nel sangue materno, diventando rilevabili già dalla 5ª settimana di gestazione. La loro concentrazione aumenta progressivamente e scompaiono dopo il parto. L'analisi di questo DNA fetale libero circolante, isolato dal sangue materno, avviene tramite sequenziamento massivo parallelo dell'intero genoma fetale, dimostrando un'attendibilità superiore al 99% nella rilevazione delle aneuploidie cromosomiche comuni (trisomia 21, 18, 13, monosomia X), con percentuali di falsi positivi inferiori allo 0,1%.
Altre Trisomie Comuni e Sindromi da Microdelezione
Le anomalie cromosomiche possono coinvolgere altri cromosomi, oltre al 16. Tra le più note vi sono:
- Trisomia 21 (Sindrome di Down): Causa genetica più comune di ritardo mentale, caratterizzata dalla presenza di un cromosoma 21 in più. Colpisce circa 1 neonato su 700. La disabilità cognitiva e la crescita fisica sono ritardate, e vi è una maggiore predisposizione a specifiche patologie. Il grado di disabilità varia ampiamente tra gli individui affetti.
- Trisomia 18 (Sindrome di Edwards): La presenza di un cromosoma 18 in eccesso è associata a un’elevata abortività e a grave ritardo mentale. I neonati affetti spesso presentano difetti cardiaci congeniti e altre condizioni patologiche che riducono significativamente l’aspettativa di vita.
- Trisomia 13 (Sindrome di Patau): Simile alla trisomia 18 per elevata abortività e grave ritardo mentale. Neonati con trisomia 13 manifestano numerosi difetti cardiaci, gravi deficit cognitivi e ritardi nello sviluppo. La sopravvivenza oltre i primi mesi di vita è rara.
Le anomalie legate ai cromosomi sessuali (X e Y) non causano generalmente deficit cognitivi o dello sviluppo fisico-motorio gravi come le trisomie autosomiche. La diagnosi precoce è fondamentale per fornire ai bambini i servizi necessari a raggiungere il loro potenziale massimo. Esempi includono la Sindrome di Turner (Monosomia X) nelle femmine, la Sindrome di Klinefelter (XXY) nei maschi, la Sindrome della tripla X (XXX) e la Sindrome di Jacobs (XYY).
Le sindromi da microdelezione, invece, sono caratterizzate dalla perdita di piccoli tratti cromosomici e dei geni in essi contenuti. Esempi significativi includono:
- Sindrome di Di George: Causata da una microdelezione sulla regione 22q11.2, si manifesta con ipoplasia del timo e delle ghiandole paratiroidi, cardiopatie congenite e caratteristici dismorfismi facciali.
- Sindrome Cri-du-chat: Dovuta alla delezione di una porzione del braccio corto del cromosoma 5 (5p-), presenta un pianto acuto e monotono (simile al miagolio di un gatto), microcefalia, tratti facciali distintivi e grave ritardo psicomotorio e mentale.
- Sindrome di Prader-Willi e Sindrome di Angelman: Condizioni neurologiche complesse che, sebbene abbiano meccanismi genetici diversi (imprinting genomico), sono associate a disabilità intellettiva, dismorfismi facciali e ritardo dello sviluppo.
- Sindrome da delezione 1p36: Caratterizzata da dismorfismi facciali tipici, ipotonia, ritardo dello sviluppo, deficit cognitivo, convulsioni e anomalie cardiache, sordità e ritardo della crescita.
- Sindrome di Wolf-Hirschhorn: Determinata da una delezione sul braccio corto del cromosoma 4 (regione 4p16.3), presenta segni craniofacciali caratteristici, ritardo della crescita, deficit cognitivo, grave ritardo psicomotorio, convulsioni e ipotonia. La sua prevalenza è di circa 1:50.000 nati.
La trisomia 16q distale, un'altra rara anomalia cromosomica causata dalla trisomia parziale del braccio lungo del cromosoma 16, presenta un quadro clinico variabile dominato da ritardo dello sviluppo, grave disabilità intellettiva, ipotonia, dismorfismi facciali specifici (fronte alta e prominente, epicanto, orecchie displastiche, sella nasale infossata, ipoplasia malare, palato stretto, micrognazia) e anomalie degli arti (aracnodattilia, piedi equino-vari). Possono associarsi cardiopatie, malformazioni genito-urinarie e anomalie vertebrali.
Prevenzione e Consulenza Genetica
La prevenzione primaria in ambito genetico si traduce nell'informazione. La figura del Genetista assume un ruolo indispensabile nella consulenza genetica, finalizzata a spiegare e interpretare problemi cromosomici al paziente e ai suoi familiari.
Le informazioni fornite in questo articolo non costituiscono in alcun modo consulenza medica e sono presentate a scopo puramente informativo, potendo non essere del tutto accurate in ogni contesto specifico.

La consulenza genetica è fondamentale anche dopo la diagnosi di un'anomalia cromosomica in un bambino. In questi casi, è raccomandato sottoporre i genitori a test genetici. Se uno dei genitori risulta essere portatore della stessa duplicazione o alterazione, il rischio di trasmetterla in future gravidanze aumenta significativamente, attestandosi al 50% in ogni nuova gravidanza. La prevenzione secondaria si basa quindi su un’adeguata consulenza genetica e sul ricorso a test diagnostici appropriati, laddove indicati, per aiutare le coppie a prendere decisioni informate riguardo alla pianificazione familiare.
È importante sottolineare che non esiste una cura risolutiva per le duplicazioni del cromosoma 16 o altre anomalie cromosomiche strutturali, poiché non è possibile rimuovere il materiale genetico in eccesso o alterato da ogni cellula dell’organismo. Tuttavia, terapie mirate e programmi educativi strutturati, come l'analisi applicata del comportamento (ABA) per bambini con diagnosi di autismo o ADHD, possono migliorare significativamente la qualità della vita e il potenziale di sviluppo degli individui affetti. La prognosi per individui con duplicazioni del cromosoma 16 è estremamente variabile e dipende in larga misura dalla tempestività e dall'efficacia degli interventi terapeutici e riabilitativi precoci.