La Sindrome di Williams (SW) è una rara condizione genetica multisistemica del neurosviluppo che colpisce individui di entrambi i sessi, caratterizzata da un insieme complesso di manifestazioni che spaziano dalle peculiarità facciali a cardiopatie, disabilità intellettive e tratti comportamentali distintivi. Comprendere a fondo questa sindrome è fondamentale per fornire un supporto adeguato ai pazienti e alle loro famiglie, navigando le sfide mediche, sociali ed emotive che essa comporta.
Origini Genetiche e Meccanismi Molecolari
La Sindrome di Williams è causata da una microdelezione sul braccio lungo (q) del cromosoma 7, specificamente nella regione 15q11-q13. Questa delezione implica la perdita di circa 26-28 geni, tra cui il gene ELN (elastina), la cui carenza è direttamente correlata alle anomalie cardiovascolari tipiche della sindrome, in particolare la stenosi sopravalvolare aortica. La prevalenza della sindrome è stimata intorno a 1:7.500 nascite, con una frequenza leggermente inferiore rispetto a quanto inizialmente ipotizzato in alcuni studi (1:134.117 contro una stimata di 1:10.000-1:52.000).
La sindrome può essere ereditata da un genitore affetto, con una probabilità del 50% di trasmissione ai figli. Tuttavia, nella maggior parte dei casi (circa l'80%), la sindrome insorge spontaneamente (de novo) durante la formazione dei gameti o nelle prime fasi dello sviluppo embrionale. Questo significa che, a meno che non ci sia una storia familiare nota, il rischio di ricorrenza per i genitori di un bambino con SW è generalmente basso (<1%). In casi rari, è stata documentata la trasmissione da un genitore affetto, il quale potrebbe presentare una forma più lieve della sindrome o la delezione potrebbe essere ereditata dal padre.
La diagnosi genetica si avvale di tecniche molecolari specifiche. La FISH (Fluorescent in situ hybridization) è una metodica che consente di visualizzare la presenza o l'assenza di specifiche sequenze di DNA sui cromosomi, identificando la microdelezione sul cromosoma 7. Test più avanzati come la PCR a metilazione specifica (MS-PCR) e il Southern blot possono fornire ulteriori dettagli. In alcuni casi, i risultati possono essere confermati mediante analisi dei microsatelliti o test di metilazione, che risultano positivi nel 98% dei pazienti.
Manifestazioni Cliniche: Un Quadro Multisistemico
Le manifestazioni cliniche della Sindrome di Williams sono estremamente variabili e possono presentarsi con diversa gravità in ciascun individuo. Tuttavia, alcuni tratti sono particolarmente caratteristici:
Dismorfismi Facciali
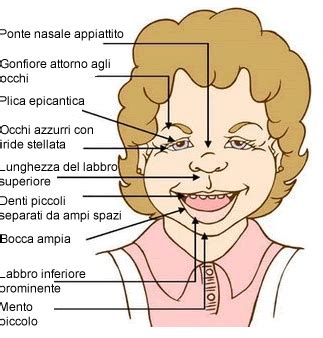
Le peculiarità facciali sono quasi universalmente presenti (nel 100% dei pazienti) e cambiano con l'età. Nei bambini piccoli si osservano tratti come l'epicanto (una piega cutanea che copre l'angolo interno dell'occhio), guance piene, un profilo facciale piatto, denti piccoli e iperdistanziati, e un'iride dall'aspetto "a stella". Nei bambini più grandi e negli adulti, il viso tende a diventare più stretto, con un collo allungato e labbra carnose con eversione del labbro inferiore. Il naso è spesso corto con narici anteverse (all'insù) e il filtro (la zona tra il naso e il labbro superiore) appare lungo.
Anomalie Cardiovascolari
Le malattie cardiovascolari sono una complicanza frequente, interessando circa l'80% dei pazienti. La stenosi sopravalvolare aortica è la più comune (70%), ma si possono riscontrare anche stenosi delle arterie di medie e grandi dimensioni, ipertensione e degenerazione dei lembi valvolari aortici e/o mitralici. Le stenosi cardiovascolari possono progredire, specialmente nei primi 5 anni di vita, rendendo essenziale un attento monitoraggio cardiologico, soprattutto nel primo anno di vita. Le stenosi polmonari periferiche tendono invece a risolversi spontaneamente.
Sviluppo Cognitivo e Comportamentale
La disabilità intellettiva è comune (75%), con un profilo cognitivo caratteristico. I pazienti con SW mostrano una relativa forza nel linguaggio e nella memoria verbale a breve termine, ma una significativa debolezza nelle capacità visuo-spaziali e motorie. Contrariamente ad altre sindromi con disabilità intellettiva, i soggetti con Williams hanno una forte predisposizione alle interazioni sociali, mostrando una personalità molto amichevole e socievole. La passione per la musica è un tratto distintivo, accompagnata però da un'ipersensibilità ai suoni (90%).
La disregolazione emotiva è frequente, e molti pazienti (50%) necessitano di trattamento farmacologico per ansia e/o deficit di attenzione/iperattività. Possono manifestare bugie e, occasionalmente, furti, evidenziando una limitata capacità di gestire i rischi e le situazioni pericolose.
Anomalie Endocrine
Diverse anomalie endocrine possono essere presenti:
- Ipercalcemia: Rilevata nel 15-45% dei casi, richiede un attento monitoraggio del calcio sierico nei primi 2 anni di vita.
- Intolleranza al glucosio e Diabete Mellito di Tipo 2: L'obesità è comune negli adolescenti e negli adulti, spesso associata a un eccessivo apporto calorico.
- Ipotiroidismo Subclinico: Presente nel 15-30% dei pazienti.
- Osteopenia/Osteoporosi: Rilevate nel 50% dei casi.
Ipotonia e Problemi Motori
L'ipotonia assiale (riduzione del tono muscolare generale) è una caratteristica neonatale marcata, che causa difficoltà nell'alimentazione e nel controllo respiratorio. Questa ipotonia centrale, associata a ipertonia periferica con aumento dei riflessi tendinei profondi negli arti inferiori, atassia e tremore, contribuisce al ritardo dello sviluppo motorio. Nei bambini piccoli si osserva lassità articolare, mentre nei più grandi e negli adulti possono svilupparsi contratture articolari che determinano un'andatura goffa. Lordosi, cifosi e scoliosi sono frequenti, con la necessità di un attento monitoraggio e, in alcuni casi, di interventi chirurgici.
Altre Manifestazioni
- Problemi Renali e Genito-Urinari: La valutazione per possibili malformazioni congenite dell'apparato genito-urinario è parte integrante del protocollo di controllo. Negli uomini, si possono riscontrare criptorchidismo (testicoli non discesi) e pene o scroto di dimensioni ridotte.
- Problemi Oculari: Anomalie oculari come strabismo, ipermetropia e astigmatismo sono comuni.
- Problemi Gastrointestinali: Difficoltà nell'alimentazione, reflusso gastroesofageo e stipsi sono frequenti nei primi mesi di vita. La crescita nei bambini è generalmente ridotta, attestandosi intorno al 75% del tasso di crescita normale.
Diagnosi Prenatale e Screening
Il Servizio di Diagnosi Prenatale e Medicina Fetale offre un percorso clinico-diagnostico completo per il monitoraggio della gravidanza e la valutazione del benessere fetale. Questo include:
- Ecografia Precoce (6-10 settimane): Valuta l'evolutività della gravidanza, la datazione, l'impianto e la corionicità in caso di gravidanze gemellari. Nel caso di gravidanze monocoriali, viene offerto un monitoraggio specifico per la Sindrome da Trasfusione Feto-Fetale (TTTs).
- Screening per Aneuploidie nel I Trimestre (Test Combinato/Bi-Test): Eseguito tra 11-13+6 settimane, combina valutazioni ecografiche (come la misurazione della translucenza nucale) con dosaggi ematici materni (fBhCG, PAPP-A). Questo test ha un'elevata sensibilità (95-97%) nel rilevare il rischio di patologie cromosomiche e può identificare spie di rischio per malformazioni fetali.
- Screening della Preeclampsia: Valuta il rischio di sviluppare preeclampsia, una complicanza caratterizzata da ipertensione e proteinuria, che può portare a ritardo di crescita fetale e parto pretermine. Lo screening, eseguito nel primo trimestre e ripetuto nei trimestri successivi, combina dati anamnestici, ecografici (Doppler delle arterie uterine), clinici (pressione arteriosa) e laboratoristici (PAPP-A, PLGF, sFLT-1). In caso di rischio elevato, è possibile iniziare un trattamento preventivo con aspirina a basso dosaggio.
- Test di Screening Non Invasivo del DNA Fetale (NIPT): Un prelievo di sangue materno che valuta il rischio di patologie cromosomiche, proposto in particolare per rischi intermedi o per una maggiore serenità della coppia.
- Ecografia di Screening del II Trimestre (Ecografia Morfologica, 19-23 settimane): Un esame dettagliato volto a identificare malformazioni congenite degli organi e apparati fetali.
- Ecocardiografia Fetale: Eseguita in caso di sospetto di cardiopatia o familiarità, per uno studio approfondito del cuore fetale.
- Ecografia di Screening del III Trimestre (30-34 settimane): Monitora l'accrescimento fetale, il benessere, la funzionalità placentare e gli scambi materno-fetali tramite flussimetria Doppler.
Qualora gli screening rilevino un rischio dubbio o aumentato, vengono offerte procedure di diagnostica prenatale invasiva come la villocentesi (11-14 settimane) o l'amniocentesi (da 16 settimane in poi) per ottenere un cariotipo fetale preciso. La villocentesi, in particolare, permette una diagnosi genetica precoce con un rischio di aborto sovrapponibile a quello dell'amniocentesi, soprattutto se eseguita da operatori esperti.
Gestione e Terapie
La gestione della Sindrome di Williams richiede un approccio multidisciplinare e personalizzato, che accompagni il paziente lungo tutto l'arco della vita.
Supporto Nutrizionale Neonatale
L'ipotonia neonatale marcata e le difficoltà di suzione rendono spesso necessario un supporto nutrizionale intensivo. Molti neonati con SW richiedono un'alimentazione enterale mediante sondino naso-gastrico per diversi mesi, fino a quando non raggiungono una crescita adeguata e una maggiore autonomia nell'alimentazione. Un aumento della densità delle poppate può essere utile. È fondamentale un monitoraggio frequente del peso e della crescita, con l'obiettivo di raggiungere un incremento ponderale adeguato.
Terapia Ormonale
La terapia con ormone della crescita ricombinante (rhGH) è stata studiata per migliorare la crescita nei bambini con SW. Alcuni studi indicano una risposta significativa, sebbene la dose e la durata del trattamento debbano essere attentamente valutate.
Gestione delle Comorbilità
- Cardiologia: Follow-up cardiologico scrupoloso, con particolare attenzione alla progressione delle stenosi.
- Endocrinologia: Monitoraggio del calcio sierico, della tolleranza al glucosio e della funzione tiroidea. Gestione dell'obesità tramite programmi nutrizionali e di attività fisica.
- Ortopedia: Monitoraggio e gestione della scoliosi e di altre problematiche scheletriche. La valutazione con bending test è raccomandata ad ogni visita di controllo.
- Respiratorio: La sindrome da apnea ostruttiva nel sonno è una complicanza che richiede attenzione. La valutazione con bending test è utile per identificare potenziali compromissioni respiratorie.
- Riabilitazione: Fisioterapia, terapia occupazionale e logopedia sono essenziali fin dalla prima infanzia per supportare lo sviluppo motorio, le abilità pratiche e la comunicazione.
Supporto Comportamentale e Psicologico
La gestione dei disturbi del comportamento, dell'ansia e dell'iperattività richiede un approccio comportamentale strutturato, spesso con il supporto di neuropsichiatri infantili e psicologi. L'obiettivo è stabilizzare il disturbo del comportamento, promuovere l'autonomia e migliorare le interazioni sociali. Un approccio graduale, con obiettivi a breve termine che portino a traguardi più ampi, è spesso efficace.

Prospettive Future e Ricerca
La ricerca continua a esplorare nuove strategie terapeutiche e a migliorare la comprensione della Sindrome di Williams. Progetti innovativi, focalizzati sul potenziamento delle abilità sociali e sull'aumento della fiducia in sé stessi, rappresentano un passo fondamentale per migliorare la qualità della vita dei pazienti. L'obiettivo è non solo fornire cure mediche, ma anche supportare pienamente l'individuo nel raggiungimento del suo potenziale.
L'aspettativa di vita per i soggetti con Sindrome di Williams non è ancora stata definita con precisione, ma le complicanze cardiovascolari rappresentano la principale causa di mortalità. Tuttavia, con una gestione medica ottimale e un supporto continuo, è possibile migliorare la prognosi e garantire una vita più piena e soddisfacente.
Conclusioni Parziali
La Sindrome di Williams è una condizione complessa che richiede un impegno congiunto da parte di medici, terapisti, famiglie e pazienti stessi. La diagnosi precoce, un monitoraggio attento delle diverse manifestazioni cliniche e un piano di trattamento individualizzato sono le chiavi per affrontare efficacemente le sfide poste da questa sindrome. La speranza risiede nella continua ricerca e nell'avanzamento delle conoscenze, che permettono di avvicinarsi sempre più a traguardi prima impensabili, proprio come il viaggio verso Marte che oggi appare più vicino rispetto al passato.
tags: #sindrome #williams #maschi #stati #uniti #amniocentesi