La Sindrome di Rett (RTT) è un disturbo neuroevolutivo genetico raro, che colpisce quasi esclusivamente le femmine e rappresenta la seconda causa di disabilità intellettiva negli individui di sesso femminile. È una malattia rara, la cui incidenza è stimata attorno a 1 ogni 10.000 nati. La sua complessità e la variabilità dei sintomi rendono la diagnosi una sfida, spesso ritardata di anni, come dimostrano drammaticamente alcune esperienze personali. Questo articolo esplora la natura della Sindrome di Rett, le sue basi genetiche, il percorso diagnostico attuale e futuro, e risponde in modo approfondito alla domanda cruciale sulla possibilità di rilevarla tramite amniocentesi.

Una Storia di Diagnosi Ritardata: Il Percorso di Claudia e Marina
La ricerca di una diagnosi precisa per una malattia rara può trasformarsi in un'odissea lunga e frustrante, un percorso che per Marina Cometto, madre di Claudia, è durato ben 38 anni. Questa è la storia di una battaglia contro l'ignoto, conclusasi con l'identificazione della Sindrome di Rett grazie all’esame del DNA effettuato attraverso una semplice analisi del sangue. La scoperta è stata un fulmine a ciel sereno per Marina: dopo aver contattato la mamma di un'altra bambina affetta da RTT, ha deciso di sottoporre sua figlia a un test che ha confermato la sua ipotesi, la presenza di una malattia rara che per 38 anni nessun medico aveva mai diagnosticato. Si è rivolta al laboratorio genetico di Siena, dove hanno ritenuto verosimile che Claudia fosse affetta da RTT. Successivamente, il test diagnostico è stato effettuato all’ospedale Molinette di Torino, che ha dato esito positivo.
Marina racconta la sua esperienza, sperando che non si ripeta. "Durante il parto," spiega Cometto, "ho avuto delle complicazioni perché mia figlia si presentava di faccia ed è nata cianotica. Secondo i medici, questo è stato il motivo delle gravi limitazioni che nel corso degli anni si sono presentate." Dopo la nascita, Claudia è stata dimessa il sesto giorno, come era solito fare allora. La piccola era ancora violacea ma mangiava, solo si stancava presto e durante la poppata era necessario interrompersi per farla riposare. A 2 mesi emetteva suoni gioiosi, a 3 mesi aveva il controllo del capo, a 4 mesi teneva in mano correttamente i giochi dei neonati, a 5 mesi è iniziato lo svezzamento. Era una bambina sempre sorridente ma la posizione da seduta, che normalmente si acquisisce verso i 6 mesi, lei l’ha conquistata a 9 mesi.
La preoccupazione di Marina è iniziata presto. "Il fatto che a 6 mesi non avesse ancora acquisito la posizione seduta mi ha preoccupato subito (facevo il confronto con la sorella maggiore) ma il pediatra mi diceva che non tutti i bambini sono uguali, che ognuno matura in modo diverso. Dovevo avere pazienza e anche Claudia avrebbe fatto tutte le cose che facevano i suoi coetanei." Ha iniziato a camminare a 18 mesi, diceva qualche parola, gironzolava per casa come tutti i bimbi. Poi ha iniziato a perdere le capacità acquisite: ha smesso di dire quelle poche parole, piano piano la coordinazione dei movimenti si è fatta più critica, cadeva spesso, era più instabile sulle gambine, fino a che ha dovuto essere sostenuta per muovere qualche passo. Ha smesso di usare le mani con movimenti volontari; le sue manine “sfarfallavano” e “sfarfallano” tutt’ora, ma senza coordinazione. Solo gli occhi hanno mantenuto un vivace contatto con ciò che la circonda e il sorriso è la sua manifestazione per dirci che sta bene. Ha anche smesso di piangere, il dolore lo manifesta irrigidendosi e lamentandosi.
Ora che Claudia ha una diagnosi, per Marina è cambiato molto, anche se le limitazioni causate dalla sindrome non saranno purtroppo reversibili. "Ora almeno quando dico il nome della patologia," spiega Cometto, "i medici si rendono conto della gravità della situazione. Prima si parlava genericamente di cerebropatia, che vuol dire tutto e niente allo stesso tempo. Ora sappiamo che le cellule cerebrali non sono distrutte per lesioni durante la nascita ma sono immature, hanno interrotto la loro crescita e questo può forse dare un po’ di speranza in più nella ricerca di una possibile cura. Grazie alla diagnosi stiamo facendo una cura per l’osteoporosi precoce che ha Claudia e che è una prerogativa della sindrome di Rett."
La figlia di Marina è ora seguita presso il Centro di neuropsichiatria infantile presso il Policlinico Le Scotte di Siena, diretto dal dott. Hayek, per sottoporsi a specifici esami neurologici, cardiaci e respiratori. Presso questo centro Marina ha trovato un punto di riferimento e la possibilità di aiutare Claudia a stare meglio. Questa esperienza sottolinea l'importanza di una diagnosi tempestiva e accurata, non solo per la comprensione della malattia, ma anche per accedere a cure di supporto specifiche e migliorare la qualità di vita del paziente.

La Sindrome di Rett: Inquadramento Nosografico, Epidemiologia e Fenotipo Clinico
La Sindrome di Rett (RTT) è un disturbo dello sviluppo di tipo neurologico che colpisce principalmente, ma non esclusivamente, soggetti di sesso femminile. Deve il suo nome al medico austriaco Andreas Rett, che per primo la riconobbe nel 1966, in seguito alla casuale osservazione nel suo ambulatorio di due bambine che avevano un simile movimento delle mani, come se le stessero lavando. Rivalutò altre bambine che aveva visitato in precedenza e pubblicò il primo articolo in cui si definiva la malattia. Tuttavia, si è dovuto attendere il 1983 e una seconda pubblicazione (a firma del ricercatore svedese Dr. B. Hagberg) perché la sindrome acquisisse riconoscimento clinico internazionale. Nel DSM-IV era classificata tra i disturbi generalizzati dello sviluppo, spesso sotto PDD-NOS, mentre nel DSM-5 e DSM-5-TR è inclusa tra i disturbi dello spettro autistico con nota esplicativa sulla genesi genetica, evidenziando le differenze fenotipiche rispetto all’ASD classico.
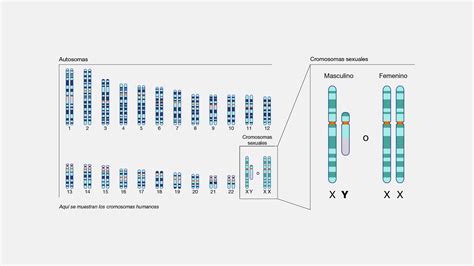
La RTT è una malattia genetica rara con un’incidenza stimata di 1 su 10.000-15.000 femmine nate vive. La RTT classica colpisce quasi esclusivamente femmine, a causa della localizzazione del gene MECP2 sul cromosoma X. I maschi con mutazioni de novo spesso sviluppano quadri neurodegenerativi gravi e non sopravvivono oltre i primi anni di vita, a meno di anomalie cromosomiche complesse o mosaicismi. L’aumento dei casi diagnosticati negli ultimi decenni riflette il miglioramento delle tecniche genetiche, una maggiore consapevolezza clinica e linee guida pediatriche più dettagliate. Studi multicentrici hanno evidenziato che l’età media di riconoscimento della RTT è tra 18 e 30 mesi, con ritardi più frequenti nelle forme atipiche.
La sindrome di Rett: cos’è e quanto è importante la ricerca in questo campo
I sintomi della sindrome di Rett nella forma classica si manifestano dopo uno sviluppo prenatale e postnatale che appare normale fino a circa 6-18 mesi, a volte anche 36 mesi, quando si arresta l’acquisizione delle tappe dello sviluppo. Questo arresto è seguito da una regressione dello sviluppo, un elemento tipico che può indirizzare i medici a sospettare la diagnosi. Durante questa fase di regressione, i bambini perdono le abilità precedentemente acquisite, come l’uso intenzionale delle mani e il linguaggio verbale. La disabilità diventa evidente con la comparsa di movimenti stereotipati delle mani, come torcere, battere o il gesto di lavarle. Si assiste anche a un comportamento ritirato, definito come "autistico". Tuttavia, questo atteggiamento viene poi recuperato nel tempo e anche se la maggior parte delle bambine con sindrome di Rett non sono in grado di parlare, non perdono il contatto visivo e comunicano con lo sguardo, molto intenso. È per questa ragione che vengono definite come le “bimbe dagli occhi belli”.
La deambulazione può essere conservata più a lungo, spesso non viene persa, anche se con la crescita può comparire una severa scoliosi che può invalidare anche l’autonomia del cammino. Un’altra problematica di rilievo nella sindrome di Rett è l’epilessia, in alcuni casi anche farmaco-resistente, ossia non controllabile con farmaci antiepilettici, e la presenza di apnee e iperventilazione. A tutto ciò si accompagna il rallentamento della crescita della circonferenza cranica, che viene definita microcefalia, e una scarsa crescita del corpo, per cui le bambine sono spesso molto magre e piccoline. La Sindrome di Rett è caratterizzata da ampia eterogeneità clinica e, oltre alla forma classica, sono note delle forme varianti, che differiscono nel periodo di esordio e nel tipo di severità clinica.

Eziologia Genetica e Meccanismi Neurobiologici Sottostanti
La Sindrome di Rett (RTT) è principalmente una malattia genetica monogenica legata al cromosoma X. Il gene MECP2 (Methyl-CpG-binding protein 2), localizzato in Xq28, è il principale responsabile della RTT classica, identificato nel 1999. MECP2 codifica per una proteina regolatrice che lega DNA metilato e controlla l’espressione genica, la cromatina e lo sviluppo neuronale. Se il gene è mutato, la proteina MeCP2 non viene prodotta o comunque viene prodotta in una forma che funziona in modo non corretto.
La maggior parte dei casi sono dovuti a mutazioni de novo nel gene MECP2, principalmente in femmine. Poiché nei maschi è presente un solo cromosoma X, un difetto in MECP2 ha un effetto ancora più devastante fino a essere letale, mentre nelle bambine la presenza di una copia sana e una difettiva del gene determina la manifestazione della sindrome di Rett. Si spiega così perché la sindrome colpisca principalmente le persone di sesso femminile.
Tuttavia, ad oggi solo in circa l’80% delle pazienti con diagnosi di sindrome di Rett classica è possibile mettere in evidenza una mutazione nel gene MECP2. Questa percentuale è molto più bassa se vengono considerate le varianti della sindrome. Oltre alla forma classica, sono state infatti descritte almeno 5 varianti. Molti studi sono stati indirizzati e sono tuttora volti a comprendere il ruolo del gene MECP2 nella sindrome. Si è dimostrato che il gene MECP2 è un regolatore della trascrizione, ovvero un gene che coordina altri geni. Si può quindi considerare come un capo operaio in una grande fabbrica. Quali siano i geni regolati da MECP2 non è ancora del tutto chiaro.
Grazie a uno studio collaborativo, è stato identificato un nuovo gene responsabile della variante con convulsioni ad esordio precoce, il gene CDKL5. Si è dimostrato che il gene CDKL5, se alterato, causa la variante con convulsioni ad esordio precoce della sindrome di Rett (Scala, E. et al. J Med Genet. 2005 Feb;42(2):103-7). Tale variante è stata per la prima volta descritta da Hanefeld nel 1985, in una bambina con spasmi infantili e ipsaritmia, che più tardi ha sviluppato la sindrome di Rett. In questa variante non sono mai state riportate mutazioni del gene MECP2. La proteina CDKL5, come la proteina MeCP2, è espressa nel cervello, ovvero i due operai lavorano nello stesso settore della fabbrica. Si è chiarito qual è il compito dell’operaio CDKL5 e dimostrato che i due operai lavorano insieme, e che, in determinate circostanze, interagiscono direttamente.
Esiste infine un terzo gene, FOXG1, che non si trova sul cromosoma X e in questo caso la malattia colpisce con uguale frequenza entrambi i sessi. I pazienti con mutazione in FOXG1 presentano una forma congenita della sindrome di Rett, che ha manifestazioni cliniche molto severe.
La neurobiologia della Sindrome di Rett riflette le conseguenze dirette delle mutazioni nel gene MECP2, che alterano la regolazione trascrizionale, la plasticità sinaptica e lo sviluppo corticale. Studi molecolari, elettrofisiologici e di neuroimaging hanno chiarito molteplici livelli di disfunzione neuronale, evidenziando schemi di connettività, trasmissione sinaptica e network cerebrali alterati. Studi post-mortem e modelli murini mostrano una ridotta densità di spine dendritiche corticali e disorganizzazione delle sinapsi glutammatergiche, associate a deficit cognitivi e motori. Alterazioni del bilancio GABA/glutammato sono documentate in diverse regioni corticali e limbiche, contribuendo a iperexcitabilità neuronale e vulnerabilità a epilessia. La perdita di MECP2 riduce il BDNF (brain-derived neurotrophic factor), compromettendo la sopravvivenza e la maturazione neuronale.
Il Percorso Diagnostico: Dalla Sospensione Clinica alla Conferma Genetica
La diagnosi della Sindrome di Rett si basa su un attento processo clinico, che include l’anamnesi familiare e l'osservazione dei criteri diagnostici principali. Questi criteri includono la perdita regressiva di abilità acquisite come linguaggio, destrezza manuale e abilità motorie fini, tipicamente tra 6 e 18 mesi di età. Altri segni distintivi sono le stereotipie manuali, come movimenti ripetitivi delle mani (torcere, battere o lavare), la decelerazione della crescita cranica e alterazioni motorie quali rigidità, atassia e problemi di equilibrio. La compromissione dello sviluppo sociale e comunicativo, con riduzione del contatto oculare e perdita di gesti intenzionali, è anch'essa significativa.
La diagnosi definitiva viene fatta in laboratori di genetica dedicati mediante un test di “sequenziamento del DNA” che ha lo scopo di leggere la sequenza del gene e verificare la presenza di errori nel gene MECP2 o in altri geni correlati come CDKL5 e FOXG1. È importante sottolineare che la diagnosi genetica mediante identificazione di mutazioni MECP2 permette conferma clinica e differenziazione dai disturbi dello spettro autistico tradizionali, pur condividendo alcune caratteristiche comportamentali.
La diagnosi precoce è essenziale per l’intervento tempestivo. L'esperienza di Marina Cometto evidenzia le sfide di una diagnosi differenziale, dove la RTT è stata inizialmente confusa con una generica "cerebropatia". È fondamentale che i medici siano più attenti al sintomo primario, che è la regressione delle capacità acquisite, per evitare ritardi diagnostici significativi. Un progetto coordinato dall’Azienda ospedaliero-universitaria Senese (Aous) sta lavorando per realizzare uno strumento che, grazie agli algoritmi dell’intelligenza artificiale, riconosca i neonati a rischio di sviluppare la sindrome di Rett (RTT) in base alla presenza di anomalie fenotipiche, anche vaghe e lievi, al fine di anticiparne la diagnosi. L'obiettivo è definire i fattori determinanti l’ampia variabilità clinica, consentendo una gestione della patologia a misura del paziente.

Diagnosi Prenatale: Cosa Rileva l'Amniocentesi per la Sindrome di Rett?
La possibilità di diagnosticare la Sindrome di Rett (RTT) in fase prenatale è una questione di grande rilevanza per le famiglie a rischio o per quelle che hanno già avuto un figlio affetto. È possibile eseguire una diagnosi prenatale in un campione di villi coriali o liquido amniotico prelevato durante la gravidanza. Tuttavia, le capacità diagnostiche dell'amniocentesi variano notevolmente a seconda della tipologia di esame condotto.
Innanzitutto, va precisato che l’Amniocentesi Tradizionale di Base non garantisce affatto che il feto sia esente da malattie genetiche come la RTT. Essa indaga essenzialmente su quelle forme patologiche che interessano il numero e l’aspetto grossolano dei cromosomi, ad esempio le anomalie cromosomiche numeriche (come la Sindrome di Down) o strutturali di grandi dimensioni. Nulla si potrà sapere delle piccole alterazioni dei cromosomi e sull’esistenza di alterazioni dei geni in essi contenuti, come quelle che causano la Sindrome di Rett.
Esiste poi l’opzione dell’Amniocentesi con Studio Parziale del DNA, che comprende, oltre allo studio tradizionale dei cromosomi, anche la ricerca delle mutazioni principali che causano le malattie geniche più frequenti nella popolazione. Queste malattie sono in genere trasmesse da genitori portatori sani che non sanno di esserlo. Ad esempio, questo screening può rilevare condizioni come la SMA (atrofia muscolare spinale), la Fibrosi Cistica, la Sindrome di Martin Bell o X-Fragile, la Sordità Congenita da alterazione del gene GJB2 e la Distrofia Muscolare di Duchenne. Per la Sindrome di Rett, sebbene non sia esplicitamente menzionata in questa categoria generale, se una specifica mutazione familiare (nel gene MECP2, CDKL5 o FOXG1) è già nota, è tecnicamente possibile eseguire un test mirato per quella mutazione sul campione di liquido amniotico.
La vera rivoluzione diagnostica è avvenuta con l’introduzione dei panel basati sullo studio dell’esoma, la porzione del DNA che codifica per le proteine del nostro organismo. Questa è la terza e più ampia opzione, l’indagine più estesa che possa essere oggi eseguita nel liquido amniotico, utilizzando tecniche sofisticate ed estese di analisi del DNA. Tale panel è noto come amniocentesi TRIO, un approccio che consente di indagare su un numero teoricamente completo di patologie genetiche note. In questo modo si può scoprire tutto quello che è clinicamente ed eticamente lecito indagare. Questo tipo di ricerca arriva a diagnosticare tra il 50-60% delle malattie genetiche; non permette di giungere al 100% solo perché vengono escluse tutte quelle patologie estremamente rare, quelle ad origine genetica dubbia o sconosciuta oppure quelle per le quali non ci è “eticamente“ permesso di indagare. Pertanto, con l'amniocentesi TRIO o con un test genetico mirato su liquido amniotico, la Sindrome di Rett può essere diagnosticata in utero, specialmente se c'è una storia familiare della mutazione o un forte sospetto clinico.
È importante consultarsi con il proprio medico e con un genetista per decidere quali ricerche eseguire sul liquido amniotico, considerando la specifica storia familiare e le preoccupazioni individuali. La mutazione nel gene MECP2 (oppure nel gene CDKL5) insorge nel gamete di uno dei due genitori, il padre o la madre. Se la mutazione colpisce un gamete maturo (cellula uovo o spermatozoo), il rischio di ricorrenza per la coppia è zero. Nell’ovaio e nel testicolo ci sono però molte cellule che sono precursori delle cellule uovo e degli spermatozoi. Se la mutazione colpisce uno di questi precursori, è possibile che più spermatozoi o più cellule uovo portino la mutazione. Se ciò accade, due genitori sani possono generare più di una figlia affetta (mosaicismo germinale). Maggiore è l’entità del mosaicismo, maggiore è il rischio di ricorrenza. L’entità del mosaicismo non è quantificabile con metodi di routine nelle singole coppie. Pertanto, durante la consulenza genetica si fa presente alla coppia che il rischio di ricorrenza è molto basso ma non è zero. Comprendere il difetto genetico consente anche di avere un’idea sull’evoluzione della malattia, in quanto sono note delle correlazioni tra tipo di difetto genetico e la severità clinica.

Prospettive Terapeutiche e Sperimentali: Dalla Gestione Sintomatica alla Terapia Genica
Ad oggi, non esiste ancora una cura definitiva per la Sindrome di Rett. Tuttavia, la ricerca biomedica sta compiendo passi da gigante nella comprensione della malattia e nello sviluppo di terapie innovative. L’approccio attuale si concentra sulla gestione sintomatica e sul miglioramento della qualità di vita delle pazienti attraverso interventi multidisciplinari.
La terapia occupazionale può aiutare i bambini a sviluppare le necessarie abilità per il quotidiano (vestirsi, nutrirsi e praticare arti e mestieri), mentre la terapia fisica e l’idroterapia possono favorire la mobilità. Studi clinici sono in corso per trovare terapie anche non geniche che consentano di migliorare la storia clinica delle bambine. L’unica terapia per ora approvata dall’agenzia del farmaco americana, Food and Drug Administration, è il trofinetide, la cui somministrazione, secondo i dati dei trials clinici, sembra apportare un miglioramento statisticamente significativo ai sintomi clinici. Il trofinetide per ora non è commercializzato in Italia.
La ricerca di una soluzione curativa per malattie genetiche gravi come la Sindrome di Rett è un "Santo Graal" per la ricerca biomedica. La sindrome di Rett è causata da mutazioni che colpiscono il gene MECP2, situato sul cromosoma X. Questo gene produce la proteina omonima, che è presente in tutto il corpo ed è cruciale per lo sviluppo cerebrale: la sua assenza o un’espressione errata portano a gravi disabilità cognitive e motorie. Un ostacolo significativo per le terapie geniche tradizionali nel caso della sindrome di Rett è l'equilibrio delicato del gene MECP2. Se espresso troppo poco porta alla malattia, ma se raggiunge livelli troppo elevati può risultare neurotossico. È ciò che si definisce "sensibilità al dosaggio genico" (gene dosage effect), una sfida biologica non da poco.
Grazie al lavoro pionieristico del professor Stuart Cobb dell’Università di Edimburgo, insieme alla biotech americana Neurogene, è stata sviluppata una terapia genica sperimentale innovativa che promette di trasformare la gestione della malattia: si chiama NGN-401, ed è basata su una tecnologia chiamata EXACT (Expression Attenuation via Construct Tuning). EXACT è un sistema di controllo genico intelligente che utilizza microRNA per modulare l’espressione del gene MECP2. Questo consente di mantenere la quantità di proteina prodotta entro un livello terapeutico sicuro, cellula per cellula. I risultati preclinici ottenuti su modelli murini, pubblicati su Science Translational Medicine, hanno mostrato che NGN-401 è in grado di migliorare i sintomi neurologici nei topi affetti da una forma simile alla sindrome di Rett, oltre a essere ben tollerata. Neurogene ha avviato uno studio clinico di Fase I/II su piccoli pazienti, con l’obiettivo di valutare la sicurezza e l’efficacia della terapia, che viene somministrata via intracerebroventricolare, nel tentativo di massimizzare le cellule cerebrali che ricevono la terapia. Il trial è condotto negli Stati Uniti, in Australia e nel Regno Unito e prevede di arruolare un totale di 16 bambine dai 4 ai 10 anni di età.
Procedono anche le sperimentazioni condotte da Taysha Gene Therapies su TSHA-102, una terapia genica che fornisce all’organismo una versione ridotta del gene MECP2. La sua caratteristica distintiva è che, invece di utilizzare l'intera sequenza codificante di MECP2, è stata scelta una versione molto più corta, che viene somministrata tramite un'iniezione nel liquido spinale (intratecale). Taysha ha somministrato la terapia genica al primo paziente Rett della storia nella primavera del 2023, seguito da un secondo paziente adulto in autunno. Questi studi sono per ora nella fase 1, quella in cui viene valutata la sicurezza del farmaco. La riuscita della terapia genica per una patologia del neurosviluppo è una grande sfida, ma il fatto che sia già possibile pensare a un trial clinico è molto incoraggiante. Studi eseguiti sul modello animale dal gruppo inglese di Adrian Bird hanno consentito di dimostrare che se la proteina MeCP2 viene ripristinata, i sintomi della malattia possono regredire, apparentemente anche nel topo non più giovane. Questa scoperta ha acceso molte speranze e dato il via a molti progetti di ricerca.
Presso Auxologico, con il supporto dell’Associazione Italiana sindrome di Rett, sono stati generati dei modelli paziente-specifici che sono utili sia per comprendere meglio le basi della malattia, sia per sperimentare l’efficacia di potenziali molecole terapeutiche anche in collaborazione con altri Istituti di Ricerca. Il modello paziente specifico si ottiene a partire dal sangue delle bambine, i cui globuli bianchi vengono separati e riprogrammati in cellule staminali pluripotenti e successivamente differenziate in neuroni corticali. Un nuovo progetto di ricerca sulla sindrome di Rett è avviato presso l’Università di Siena, con l'obiettivo di ovviare all’ostacolo del tessuto “malato” inaccessibile, inducendo “in vitro” la differenziazione di cellule mesenchimali staminali in senso neuronale.
La sindrome di Rett: cos’è e quanto è importante la ricerca in questo campo
Il Ruolo Fondamentale delle Associazioni e la Consapevolezza Pubblica
Per le malattie rare, l’attività delle associazioni di pazienti o di genitori è fondamentale per il supporto alle famiglie. Esse vengono messe in contatto tra di loro per condividere le proprie difficoltà ed esigenze e in contatto con clinici e ricercatori per essere sempre aggiornati e partecipi di tutti gli avanzamenti clinici e scientifici.
Marina Cometto, la mamma di Claudia, desidera far conoscere il più possibile la sindrome di Rett. "So che la malattia non è di facile diagnosi, ma se i medici fossero stati più attenti al sintomo primario, che è la regressione delle capacità acquisite, forse Claudia non avrebbe avuto il tracollo fisico e intellettivo che ha invece ha limitato di molto la sua vita. Vorrei dare il mio contributo per evitare che a altre bambine fossero rubate le opportunità di una vita piena e gioiosa. Vorrei che anche i genitori fossero più informati in modo da suggerire eventualmente ai medici almeno il dubbio della diagnosi. Non è accettabile che nel terzo millennio ci si mettano 38 anni per una diagnosi così importante."
A questo scopo, Marina ha creato un opuscolo che illustra la storia di Claudia e intende far sì che venga pubblicato e distribuito a medici di base, pediatri di libera scelta, consultori e ospedali. "Sto cercando un sostegno economico per provvedere alla stampa, per poter diffondere il più possibile informazioni sulla RTT. Sto poi scrivendo un libro dove racconto le nostre vicissitudini, e sto cercando un editore. Gli eventuali diritti d’autore saranno devoluti interamente alle associazioni che si occupano della sindrome di Rett."
In Italia, l’Associazione Italiana Sindrome di Rett, AIRETT, è molto attiva e ha creato un consorzio di specialisti sia a livello nazionale che internazionale che interagiscono attivamente tra loro. Un esempio del lavoro delle associazioni è la realizzazione della piattaforma "Amelie", nata dalla collaborazione di AIRETT con la fondazione Vodafone, tecnici e ingegneri informatici, che rende possibile alle bambine la comunicazione. In parole semplici, "Amelie" è un software che si basa sul puntamento oculare e permette di gestire da un’app sul telefono un computer con un eye tracker calibrato. In questo modo le bambine Rett, dirigendo il loro sguardo, possono comunicare ed effettuare delle scelte. Oltre ad AIRETT, che è l'associazione con il maggior numero di pazienti e la più antica in Italia, ve ne sono anche altre, quali ProRett e ConRett, tutte impegnate a sostenere la ricerca e le famiglie.