Le cardiopatie congenite rappresentano una delle malformazioni più comuni alla nascita, interessando circa un neonato su cento. Sebbene metà di questi difetti non sia grave e possa essere scoperta tardi o non necessitare di trattamento, esistono forme complesse e potenzialmente letali che richiedono un intervento immediato e specialistico. Le malformazioni del cuore possono avere differenti cause, per esempio malattie virali della mamma, l'assunzione di sostanze che danno assuefazione durante la gravidanza o malattie ereditarie. Tuttavia, nella maggior parte dei bambini colpiti, la causa del vizio cardiaco resta sconosciuta. Un aspetto cruciale nella comprensione di queste patologie è l'analisi dei "shunt", ovvero passaggi anomali di sangue tra diverse sezioni del cuore o tra i grandi vasi, che possono essere vitali per la sopravvivenza o, al contrario, causa di grave compromissione emodinamica. La complessità dei difetti cardiaci congeniti è tale che le categorie diagnostiche elaborate dalla Società Europea di Cardiologia Pediatrica (AEPC) distinguono 2000 diverse manifestazioni, singole o in combinazione.
Un'associazione particolarmente significativa e ben documentata si osserva tra le cardiopatie congenite e alcune anomalie cromosomiche, tra cui spicca la Sindrome di Down, nota anche come Trisomia 21. Non è raro osservare che circa un bambino su cinque con difetti cardiaci congeniti presenta alla nascita un'ulteriore anomalia, e i bambini affetti da difetti cardiaci congeniti soffrono di frequente anche della sindrome di Down (trisomia 21). Questa forte correlazione evidenzia la necessità di uno screening cardiaco attento nei neonati con Trisomia 21 e una comprensione approfondita delle specifiche cardiopatie a cui sono più inclini.
Defetti Cardiaci Congeniti e la Sindrome di Down (Trisomia 21)
La Sindrome di Down (Trisomia 21) è un fattore di rischio estremamente rilevante per la presenza di difetti cardiaci congeniti. La relazione tra la Trisomia 21 e le cardiopatie è così marcata che circa il 40-50% dei bambini nati con questa sindrome presenta una forma di difetto del setto atrioventricolare, una delle anomalie più complesse e al contempo più frequentemente riscontrate in questa popolazione. Questa incidenza sottolinea l'importanza di una diagnosi prenatale o neonatale tempestiva delle anomalie cardiache in presenza di Trisomia 21. Tali difetti, che spesso coinvolgono la comunicazione tra le camere cardiache e le valvole, possono alterare profondamente la normale circolazione sanguigna, con conseguenze significative sulla salute e lo sviluppo del bambino. Comprendere la natura specifica di questi difetti è fondamentale per delineare percorsi diagnostici e terapeutici mirati.

I sintomi dei difetti cardiaci congeniti variano ampiamente a seconda del tipo e dell'entità del vizio. Nei lattanti, per esempio, possono manifestarsi una intensa sudorazione durante l'allattamento o l'assunzione di alimenti, una debolezza nella poppata e uno scarso aumento di peso, segni di una ridotta capacità fisica e di uno sforzo cardiaco eccessivo. Altri segnali possono includere un colorito livido di pelle e mucose, noto come cianosi, disturbi del ritmo cardiaco, disturbi respiratori, ingrossamento del fegato e problemi di crescita. Tuttavia, esistono anche difetti cardiaci che non causano alcun disturbo o solo minimi, rendendo la diagnosi più sfuggente in assenza di screening specifici. La medicina, in questo campo, non può offrire risposte generiche, data la vastità e la variabilità delle manifestazioni cliniche.
Il Difetto del Setto Atrioventricolare: Una Patologia Frequente nella Trisomia 21
Il difetto del setto atrioventricolare (DSAV) con comunicazione solo a livello atriale, noto anche come difetto del setto atrioventricolare parziale o difetto dell'ostium primum, è una malformazione cardiaca congenita che rientra nello spettro dei difetti dei cuscinetti endocardici. Questa specifica variante è un esempio calzante della stretta correlazione tra Trisomia 21 e cardiopatie, essendo presente in una percentuale significativa dei bambini affetti da questa sindrome. In questa condizione, l'anomalia anatomica principale è localizzata nella parte inferiore del setto interatriale, proprio sopra le valvole atrioventricolari, ovvero la mitrale e la tricuspide.
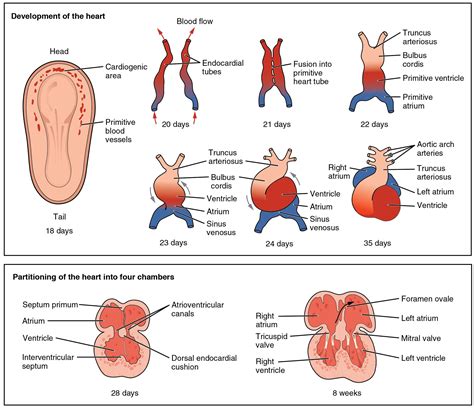
Sebbene la comunicazione sia limitata al livello atriale, la struttura delle valvole cardiache è quasi sempre coinvolta. Tipicamente, la valvola mitrale, che separa l'atrio sinistro dal ventricolo sinistro, presenta una fessura, chiamata "cleft", nel suo lembo anteriore. Questa alterazione può causare un rigurgito di sangue dal ventricolo all'atrio, complicando il quadro emodinamico del paziente e contribuendo ai sintomi associati. Dal punto di vista epidemiologico, questa condizione rappresenta una percentuale significativa delle cardiopatie congenite, rafforzando ulteriormente la sua importanza nel contesto delle anomalie cardiache.
Le cause esatte del difetto del setto atrioventricolare con comunicazione solo a livello atriale risiedono in un'alterazione dello sviluppo embrionale del cuore, che avviene tra la quarta e la settima settimana di gestazione. Durante questo periodo critico, i cuscinetti endocardici dovrebbero fondersi per formare la parte inferiore del setto atriale, la parte superiore del setto ventricolare e le valvole mitrale e tricuspide. Un'interruzione o un'anomalia in questo processo di fusione porta alla formazione del difetto. Esiste una fortissima correlazione genetica associata a questa patologia, con la Sindrome di Down (Trisomia 21) che si configura come il fattore di rischio più rilevante, influenzando fino al 40-50% dei bambini che nascono con questa sindrome e presentando una forma di difetto del setto atrioventricolare. Tuttavia, è importante notare che la forma parziale può manifestarsi anche in bambini senza anomalie cromosomiche evidenti, suggerendo che altri fattori genetici o ambientali possano contribuire alla sua insorgenza.
A differenza delle forme complete di difetto atrioventricolare, che tendono a manifestarsi precocemente nelle prime settimane di vita con sintomi evidenti, i pazienti con comunicazione solo a livello atriale possono rimanere asintomatici per diversi anni. Nei neonati e nei lattanti, tuttavia, i segni possono essere più sfumati ma pur sempre indicativi di un problema cardiaco. Un occhio attento può notare un scarso accrescimento ponderale o una sudorazione profusa durante i pasti, entrambi segni dello sforzo cardiaco compensatorio. All'esame obiettivo, il medico rileva quasi sempre un soffio cardiaco caratteristico, causato dal passaggio turbolento del sangue attraverso il difetto o dal rigurgito della valvola mitrale.
Il percorso diagnostico inizia solitamente con una visita pediatrica di routine in cui viene riscontrato un soffio cardiaco. L'ecocardiogramma è l'esame fondamentale e spesso definitivo, in quanto utilizza gli ultrasuoni per visualizzare l'anatomia del cuore in tempo reale, permettendo di identificare con precisione la localizzazione e l'estensione del difetto. L'elettrocardiogramma (ECG), pur non essendo diagnostico di per sé, in questa patologia mostra spesso un segno molto caratteristico chiamato "deviazione assiale sinistra", che aiuta a distinguere questo difetto da altri tipi di comunicazioni interatriali e fornisce informazioni aggiuntive sull'impatto emodinamico del difetto.
Il trattamento definitivo per il difetto del setto atrioventricolare con comunicazione solo a livello atriale è esclusivamente chirurgico. L'operazione viene solitamente programmata in età prescolare, tra i 2 e i 5 anni, a meno che i sintomi non siano gravi o la crescita del bambino non sia compromessa, nel qual caso si interviene prima per prevenire ulteriori danni. Durante l'intervento, la fessura (cleft) nel lembo della valvola mitrale viene suturata con precisione per ridurre o eliminare il rigurgito, e il difetto nel setto atriale viene chiuso. È importante sottolineare che i farmaci non curano il difetto anatomico, ma servono a gestire i sintomi prima dell'intervento o in caso di complicazioni post-operatorie, come l'insufficienza cardiaca. La prognosi per i pazienti sottoposti a correzione chirurgica è generalmente eccellente. La maggior parte dei bambini conduce una vita normale, può praticare sport e ha un'aspettativa di vita paragonabile a quella della popolazione generale. Tuttavia, è necessario un follow-up cardiologico a vita per monitorare la funzione cardiaca. La complicazione più comune a lungo termine è la ricomparsa o il peggioramento dell'insufficienza della valvola mitrale. In circa il 10-15% dei casi, può essere necessario un secondo intervento chirurgico anni dopo per riparare o sostituire la valvola, evidenziando la necessità di una sorveglianza continua. Poiché si tratta di una malformazione congenita legata allo sviluppo embrionale, non esiste una prevenzione certa per questa specifica anomalia.
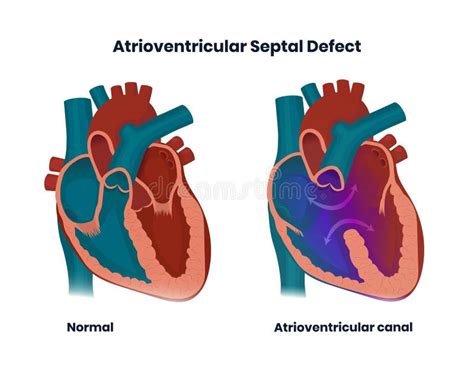
Sindrome del Cuore Sinistro Ipoplasico (HLHS): Complessità e Meccanismi
La sindrome del cuore sinistro ipoplasico (Hypoplastic Left Heart Syndrome - HLHS) è una delle più gravi forme malformative cardiache e rappresenta, al momento, una delle più grandi sfide per cardiologi e cardiochirurghi. Questa sindrome consiste in una serie di alterazioni strutturali del cuore caratterizzate da grave iposviluppo delle sezioni cardiache di sinistra, includendo l'aorta, il ventricolo sinistro e la valvola mitrale. In un cuore normale, la parte sinistra, rappresentata dall'atrio sinistro e dal ventricolo sinistro, ha la funzione cruciale di pompare sangue ossigenato in aorta e, da questa, in tutto il corpo. Nella HLHS, la compromissione di queste strutture rende impossibile questa funzione essenziale.
Il ventricolo di sinistra è costantemente ipoplasico, con vari gradi di gravità, fino all'atresia, che indica l'assenza di un lume o un'apertura della valvola. La classificazione anatomica della HLHS prevede la distinzione in quattro gruppi principali a seconda del grado di alterazione dell'aorta e della mitrale: stenosi aortica e mitralica; atresia aortica e mitralica; atresia aortica e stenosi mitralica; e stenosi aortica e atresia mitralica. Questi sottotipi evidenziano la variabilità delle presentazioni anatomiche di questa complessa sindrome.
I meccanismi di sviluppo dell'ipoplasia o atresia sinistra sembrano essere legati al fenomeno del "nessun flusso nessuna crescita" (no flow no grow), caratterizzato dalla mancata crescita delle strutture cardiache interessate da un flusso sanguigno compromesso. Questo significa che se il sangue non riesce a passare adeguatamente attraverso una valvola o una camera cardiaca durante lo sviluppo fetale, quella struttura non si sviluppa correttamente. L'incidenza della HLHS è di circa 1/5000-6000 nati vivi, rendendola una delle cardiopatie congenite più rare ma al contempo più severe.
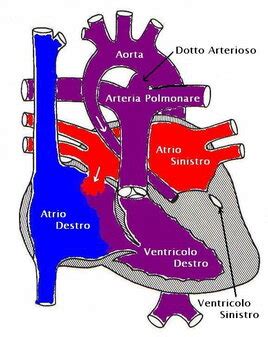
La sindrome del cuore sinistro ipoplasico classica è caratterizzata da grave ipoplasia ventricolare sinistra, grave stenosi o atresia mitralica, grave stenosi o atresia aortica e grave ipoplasia dell'arco aortico. Tuttavia, non sempre l'ipoplasia delle strutture cardiache di sinistra è tale da formulare una chiara diagnosi di sindrome del cuore di sinistra ipoplasico; in questi casi, si parla di "ventricolo sinistro borderline", indicando una situazione a rischio di progressione. Non è raro osservare, in epoca fetale, una ipoplasia ventricolare sinistra moderata che nelle fasi più avanzate della gravidanza evolve in sindrome del cuore sinistro ipoplasico, sottolineando la necessità di un monitoraggio ecocardiografico continuo.
Si distinguono due modelli di ipoplasia ventricolare sinistra, ciascuno con caratteristiche specifiche:
- Un ventricolo sinistro lungo e sottile che si associa ad arco aortico ipoplasico; in questo modello, valvola mitralica ed aortica sono spesso ipoplasiche, e la fibroelastosi endocardica, un ispessimento del rivestimento interno del cuore, in genere non è presente.
- Un ventricolo sinistro corto e spesso che si associa a grave stenosi aortica; in questo caso, si sviluppa fibroelastosi endocardica, e sono comuni anche le anomalie della valvola mitrale e l'ostruzione dell'arco aortico.
Una parte dei feti affetti da sindrome del cuore sinistro ipoplasico sviluppa una riduzione del setto atriale o la chiusura del forame ovale, una condizione denominata "sindrome del cuore sinistro ipoplasico con setto intatto". Questa situazione determina una prognosi peggiore in quanto si crea un importante ostacolo al flusso polmonare e una conseguente ipertensione polmonare, mettendo in grave pericolo la vita del neonato. In tal caso, sono spesso previsti interventi chirurgici urgenti sia in epoca prenatale che postnatale per ristabilire un flusso sanguigno adeguato. Le anomalie genetiche che possono essere associate alla HLHS includono Sindrome di Turner, Trisomia 13, Trisomia 18, Sindrome di Noonan, delezioni del cromosoma 7, Sindrome di Holt-Oram, Sindrome di Smith-Lemli-Opitz, Trisomia parziale del cromosoma 9, Sindrome di Jacobsen e altre, indicando una base genetica eterogenea per questa complessa patologia.
Il segno principe per la diagnosi di HLHS è la presenza, in sezione quattro camere all'ecocardiogramma, di una marcata discrepanza di volume tra il ventricolo di sinistra, che appare piccolo e non arriva mai a formare l'apice del cuore, e il ventricolo di destra, che è invece grande e spesso ipertrofico. Nelle forme maggiori, la valvola mitrale è atresica, e in asse lungo di sinistra l'aorta ascendente appare filiforme, indicando un grave iposviluppo. Al Color Doppler, l'atresia della mitrale comporta l'assenza del riempimento del ventricolo di sinistra ed è presente uno shunt sinistro-destro a livello del forame ovale, un meccanismo compensatorio vitale. Nelle forme con setto intatto o forame ovale restrittivo, non vi è shunt tra i due atri, e le vene polmonari appaiono spesso dilatate a causa dell'accumulo di sangue.
La Circolazione Fetale e Postnatale Nelle Cardiopatie Congenite Gravi
La circolazione sanguigna nel feto è intrinsecamente diversa da quella postnatale e si basa sulla presenza di shunt fisiologici che bypassano i polmoni, non ancora attivi, e il fegato. Nel feto con sindrome del cuore sinistro ipoplasico, l'ossigenazione dei vari organi ed apparati viene mantenuta proprio grazie a questi shunt e a un percorso circolatorio adattato. Il sangue ossigenato proveniente dalla vena ombelicale e dal dotto venoso di Aranzio viene convogliato nell'atrio destro. Da qui, gran parte di questo sangue passa nel ventricolo di destra, poi nell'arteria polmonare e da questa, attraverso il dotto arterioso di Botallo, un altro shunt cruciale, viene convogliato nell'aorta. Il sangue che arriva in aorta si divide quindi in un flusso diretto verso l'aorta discendente e quindi verso la parte inferiore del corpo, e un flusso diretto verso l'aorta ascendente e quindi i vasi del collo, la parte superiore del corpo e le coronarie. Questo percorso garantisce che anche con un cuore sinistro ipoplasico, gli organi vitali fetali ricevano sangue ossigenato.
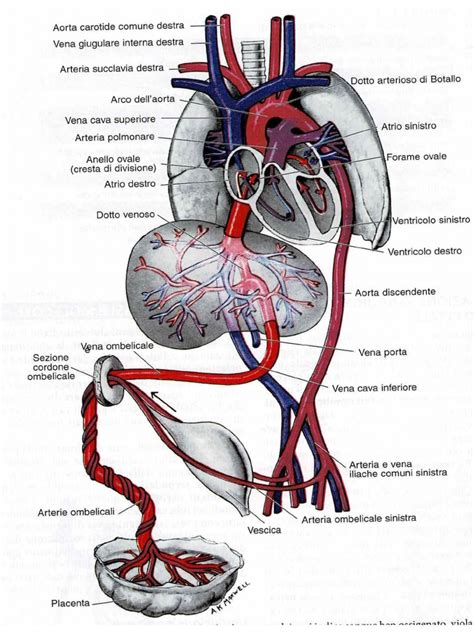
Alla nascita, con la chiusura fisiologica del dotto arterioso di Botallo, che avviene di solito durante le prime 24-48 ore di vita, e la chiusura del forame ovale, si ha il mancato passaggio di sangue verso l'aorta attraverso il percorso fetale. In un neonato con HLHS, ciò determina una situazione di crisi acuta. L'unica possibilità che ha il neonato di sopravvivere è il mantenimento della pervietà del dotto di Botallo e il mantenimento della comunicazione tra i due atri attraverso il forame ovale pervio. Quest'ultimo consente il passaggio di sangue ossigenato proveniente dalle vene polmonari dall'atrio di sinistra a quello di destra, e da qui, attraverso il percorso atrio destro >> ventricolo destro >> arteria polmonare >> dotto arterioso di Botallo, il sangue viene convogliato in aorta. Questa patologia è quindi definita "dotto-dipendente" perché la pervietà del dotto arterioso è essenziale per la sopravvivenza.
Nei casi in cui il setto interatriale è integro, con assenza della comunicazione attraverso il forame ovale, si crea un blocco significativo al flusso sanguigno, e bisogna intervenire di urgenza sia per evitare la chiusura del dotto di Botallo (spesso tramite somministrazione di prostaglandine) che per ripristinare la comunicazione tra i due atri (tramite setto-stomia atriale). Se la diagnosi di HLHS con setto interatriale intatto viene posta in epoca prenatale, si può intervenire in utero "aprendo" il setto interatriale e assicurando la persistenza della pervietà del dotto arterioso di Botallo, migliorando significativamente la prognosi.
Alla nascita, quindi, la sopravvivenza dei piccoli pazienti affetti da Sindrome del cuore sinistro ipoplasico è strettamente legata alla pervietà del dotto arterioso di Botallo e alla presenza della comunicazione tra i due atri tramite il forame ovale pervio. In alcune circostanze, è possibile la presenza di un vaso anomalo che collega la vena brachiocefalica sinistra e la vena polmonare superiore sinistra. Questa connessione alternativa, denominata Vena LevoAtrioCardinale, può consentire un drenaggio alternativo del sangue proveniente dalle vene polmonari proprio in quelle cardiopatie, come la sindrome del cuore sinistro ipoplasico, dove il cuore sinistro è malformato, offrendo un ulteriore meccanismo di compenso (Shet N. 2014).
Quando il dotto arterioso inizia a chiudersi, e ciò avviene di solito nei primi giorni di vita, diminuisce il flusso di sangue al corpo con il risultato che gli organi vitali vengono ipoperfusi, ricevendo meno sangue, e si instaura uno stato di shock, sottolineando l'estrema urgenza di mantenere la pervietà del dotto.
Altre Anomalie Cardiache Strutturali Significative
Oltre alla sindrome del cuore sinistro ipoplasico e al difetto del setto atrioventricolare, esistono altre anomalie congenite cardiache strutturali che comportano shunt o alterazioni del flusso aortico o polmonare, con rilevanza clinica significativa.
Atresia Polmonare con Setto Interventricolare Intatto (PAIVS)
L'atresia polmonare con un setto interventricolare intatto è una condizione in cui la valvola polmonare non è adeguatamente formata, impedendo così il flusso di sangue dal cuore ai polmoni. Questa anomalia si verifica frequentemente insieme all'ipoplasia della valvola tricuspide e all'ipoplasia del ventricolo destro. Questa associazione è facilmente comprensibile in base al fatto che la normale crescita ventricolare nella vita fetale dipende da un adeguato afflusso e deflusso di quel ventricolo. Senza un flusso adeguato attraverso la valvola polmonare e il ventricolo destro, queste strutture non si sviluppano pienamente.
Anomalie delle arterie coronarie, in particolare i collegamenti fistolosi delle arterie coronarie al ventricolo destro ipoplasico e stenosi delle arterie coronariche, sono comuni in PAIVS e hanno un impatto importante sulla prognosi e le opzioni chirurgiche. La chirurgia è sempre necessaria per la sopravvivenza. La sopravvivenza postnatale immediata dipende dalla pervietà del dotto arterioso, che in questo caso consente al sangue di raggiungere i polmoni. I neonati con cianosi e i rilievi auscultatori all'esordio possono comprendere un soffio da rigurgito tricuspidale o un soffio continuo dovuto alla pervietà del dotto arterioso. Se non precedentemente diagnosticata con l'ecografia prenatale, la diagnosi postnatale avviene tramite ecocardiografia, che visualizza l'assenza della valvola polmonare e le alterazioni associate.
Stenosi della Valvola Polmonare nei Bambini
La stenosi della valvola polmonare, come un reperto isolato, è un difetto congenito comune, rappresentando il 7-10% dei difetti cardiaci congeniti, e porta all'ostruzione dell'efflusso dal ventricolo destro. Questa ostruzione provoca ipertrofia ventricolare destra, poiché il ventricolo deve lavorare di più per pompare il sangue attraverso la valvola ristretta, e infine insufficienza ventricolare destra, se la condizione non viene trattata. La patologia può consistere nella fusione commissurale di una valvola a due o tre foglie o in una displasia del foglietto valvolare.
I reperti clinici comprendono un notevole click di eiezione sistolica precoce, un normale o ampio secondo tono cardiaco sdoppiato (S2) con una componente polmonare soffice e un soffio sistolico di eiezione duro, la cui intensità varia direttamente a seconda della gravità della stenosi. Questo soffio è più forte a livello del bordo superiore sinistro dello sterno. Nei neonati con stenosi polmonare critica, una cianosi è presente a causa dello shunt atriale destro-sinistro attraverso un forame ovale pervio, che permette al sangue non ossigenato di bypassare i polmoni e raggiungere la circolazione sistemica.
Il trattamento consiste solitamente nella dilatazione transcatetere con palloncino, una procedura minimamente invasiva. Se la stenosi è da moderata a grave, il trattamento chirurgico della stenosi è molto efficace, ma la chirurgia è solitamente riservata a quelle valvole che non sono suscettibili di dilatazione transcatetere con palloncino, oppure in casi di anatomia valvolare complessa.
Gamma del Ventricolo Singolo
Le anomalie del ventricolo singolo comprendono qualsiasi lesione complessa con un solo ventricolo funzionale. Questo include ipoplasia del ventricolo destro o del ventricolo sinistro e, meno comunemente, una vera camera singola ventricolare indifferenziata, dove non è possibile distinguere un ventricolo destro o sinistro separato. La tempistica della presentazione postnatale dipende da diversi fattori, tra cui la presenza di un difetto del setto interatriale, la gravità della stenosi polmonare e la pervietà del dotto arterioso. A seconda di questi elementi, i neonati possono mostrare insufficienza cardiaca o cianosi, riflettendo uno squilibrio tra il flusso sanguigno polmonare e sistemico. Se non precedentemente diagnosticata con l'ecografia prenatale, la diagnosi postnatale avviene tramite ecocardiografia, che permette di visualizzare l'anatomia complessa e la funzionalità del singolo ventricolo.
Il trattamento chirurgico per le condizioni di ventricolo singolo è un processo a più stadi che mira a creare una circolazione funzionale. Il primo obiettivo è garantire un adeguato flusso sanguigno polmonare o, al contrario, proteggere il letto vascolare polmonare dall'eccessivo flusso. Per i pazienti con flusso sanguigno polmonare ridotto, si esegue un'anastomosi sistemica all'arteria polmonare, come lo shunt modificato di Blalock-Taussig-Thomas, che devia parte del sangue dalla circolazione sistemica verso i polmoni. Se c'è sovraccarico di circolo polmonare, invece, si interviene per limitare il flusso sanguigno polmonare, ad esempio attraverso il bendaggio dell'arteria polmonare o altri interventi complessi come una ricostruzione delle radici aortiche e polmonari sul modello di quella di Norwood modificata, a volte chiamata anastomosi di Damus-Kaye-Stansel. Più tardi, la procedura di Fontan può essere utilizzata come trattamento definitivo per rendere funzionante il ventricolo singolo esclusivamente come ventricolo sistemico, separando completamente la circolazione sistemica da quella polmonare e bypassando il ventricolo destro.
Altri Difetti e Condizioni Associate
Oltre alle cardiopatie strutturali, è importante considerare anche rari difetti cardiaci non strutturali, come il blocco cardiaco completo congenito, che può causare bradicardia grave già in utero. Gli errori congeniti del metabolismo che portano alla cardiomiopatia, pur non essendo difetti strutturali primari, possono compromettere gravemente la funzione cardiaca. Anche la sindrome del QT lungo e altre sindromi genetiche aritmiche con rischi di aritmie ventricolari gravi e potenzialmente fatali, sebbene discusse altrove, fanno parte dello spettro delle anomalie cardiovascolari che possono manifestarsi in età pediatrica.
Diagnosi e Gestione delle Cardiopatie Congenite
La diagnosi precoce delle cardiopatie congenite è fondamentale per migliorare la prognosi. L'ecografia prenatale ha rivoluzionato la capacità di identificare molte di queste condizioni prima della nascita, consentendo una pianificazione accurata del parto e degli interventi postnatali. Strumenti come l'ecocardiografia fetale permettono di visualizzare in dettaglio l'anatomia e la funzione cardiaca del feto. Ad esempio, nel caso della HLHS, l'ecocardiogramma fetale può rivelare la discrepanza di volume tra i ventricoli e le alterazioni delle valvole. Per la stenosi polmonare o l'atresia polmonare con setto interventricolare intatto, l'ecocardiografia identifica l'ostruzione o l'assenza della valvola.
Dopo la nascita, l'ecocardiogramma rimane lo strumento diagnostico principe, spesso integrato da elettrocardiogrammi (ECG) e, in casi complessi, da risonanze magnetiche cardiache (RMC) o tomografie computerizzate (TC). Questi esami permettono di confermare la diagnosi, valutarne la gravità e pianificare il trattamento. I rilievi auscultatori, come i soffi cardiaci, sono spesso il primo indizio che porta a ulteriori indagini.
Le prospettive a lungo termine per i bambini con sindrome del cuore sinistro ipoplasico e altre cardiopatie congenite continuano a migliorare grazie ai progressi nella chirurgia cardiaca, nella cardiologia interventistica e nella gestione medica. La durata e la qualità della vita dipendono da molteplici fattori, tra cui la complessità del difetto, la presenza di altre anomalie associate e l'efficacia degli interventi. Sebbene oggigiorno il 90 percento dei piccoli pazienti cardiopatici raggiunga l'età adulta, la maggior parte di loro deve ricorrere per sempre a cure mediche specialistiche.
A causa della crescita del corpo e degli organi, molte terapie hanno sempre un raggio d'azione temporalmente limitato e possono richiedere interventi ripetuti nel corso della vita. È un percorso lungo che richiede un impegno costante da parte dei pazienti e delle loro famiglie. Dopo la pubertà, l'adolescente passa inoltre dalle mani fidate del cardiologo pediatrico a quelle dello specialista dell'età adulta, un passaggio delicato. E con il distacco dai genitori, il giovane cardiopatico deve assumersi consapevolmente la responsabilità della propria salute, un passo non sempre facile che richiede un'adeguata preparazione e supporto. A scuola e nella scelta della professione, il giovane affetto da difetti cardiaci congeniti si scontra talvolta con i limiti dettati dalla condizione fisica, richiedendo adattamenti e una comprensione sociale. Le anomalie extracardiache associate, che possono includere agenesia del corpo calloso, ernia diaframmatica, atresia duodenale, fistola tracheo-esofagea e onfalocele, complicano ulteriormente il quadro clinico e richiedono un approccio multidisciplinare alla cura.
Al Sant'Orsola, cardiochirurgia pediatrica: riferimento internazionale per le cardiopatie congenite
La sindrome del cuore sinistro ipoplasico o HLHS è uno dei difetti cardiaci più complessi osservati nel neonato. I difetti cardiaci sono spesso fatali senza intervento precoce, ma le prospettive a lungo termine continuano a migliorare. La diagnosi di una cardiopatia congenita è causa di grandi timori e preoccupazioni per i genitori, ma la conoscenza delle opzioni terapeutiche e un approccio multidisciplinare offrono speranza e migliorano significativamente la qualità della vita di questi bambini.