Le malattie cistiche renali rappresentano un gruppo eterogeneo di condizioni che colpiscono i reni, molte delle quali hanno origine nell'epoca fetale e sono di natura congenita. Queste patologie possono variare notevolmente per gravità, ereditarietà e impatto sulla funzione renale. Comprendere le loro specificità è fondamentale per una diagnosi precoce e una gestione ottimale.
La Displasia Renale Multicistica (MCDK): Una Malformazione Congenita Specifica
La displasia renale multicistica (MCDK), nota anche come rene multicistico, è una delle malformazioni renali più frequenti e rappresenta la patologia cistica del rene più comune, distinguendosi dalle patologie policistiche renali. Essa è caratterizzata dalla presenza di molteplici cisti, che sostituiscono il tessuto renale sano. A differenza delle patologie policistiche, che hanno una trasmissione genetica familiare e comportano una progressiva perdita di funzionalità di entrambi i reni, la displasia multicistica del rene non è a carattere ereditario e coinvolge solitamente un solo rene. La condizione di bilateralità del rene multicistico, infatti, non è compatibile con la vita.
Origine e Sviluppo della MCDK
La causa di questa patologia è da ricercare nei processi embriologici che portano allo sviluppo del rene. Durante la gestazione, la gemma ureterale, una struttura fondamentale per la formazione del rene, non raggiunge la porzione congrua del blastema metanefrico, il precursore embriologico del rene. Questo mancato raggiungimento determina un arresto nei processi di maturazione che il precursore renale dovrebbe subire, portando così alla completa assenza di tessuto funzionale al suo posto. Il rene, in queste condizioni, è completamente sostituito da cisti che non comunicano tra loro.
Nella quasi totalità dei casi, le malattie cistiche renali nel bambino, inclusa la MCDK, sono malattie congenite. La MCDK monolaterale, in particolare, esordisce spesso in epoca prenatale e viene diagnosticata in occasione delle ecografie di routine, in genere attorno alla ventesima settimana di gestazione.
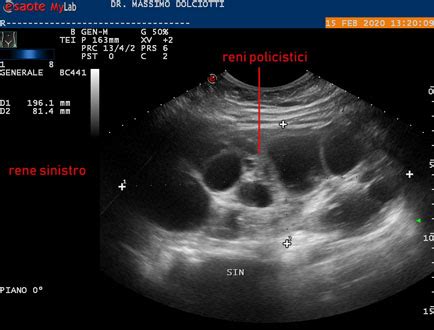
Caratteristiche Ecografiche e Diagnosi Prenatale
L'indagine strumentale principale per le malattie cistiche renali, inclusa la MCDK, è l'ecografia renale. In epoca prenatale, l'ecografia fetale a 20 settimane di gestazione permette di valutare la presenza di questa malformazione. L'ecografia evidenzia grosse cisti ipoecogene, non comunicanti, all'interno di un rene dai contorni irregolari, in assenza di un apprezzabile bacinetto renale. Il parenchima renale normale è assente o scarso. Solo nella forma segmentale è possibile visualizzare parenchima renale normale.
Per semplificare il concetto, i reni con la vescica, gli ureteri e l’uretra costituiscono una componente fondamentale dell’apparato urinario, che ha il compito di rimuovere dall’organismo le sostanze che non sono più utili, attraverso la produzione e l’eliminazione dell'urina. Nella displasia renale, il processo di formazione del o dei reni durante lo sviluppo fetale non avviene in modo corretto e l’organo risulta anormale: spesso ha dimensioni ridotte e contiene un numero inferiore al normale di nefroni, cioè le componenti fondamentali del rene, che hanno il compito di filtrare il sangue e di produrre l’urina.
Decorso Clinico e Possibili Complicanze
La maggior parte dei pazienti con MCDK monolaterale è asintomatica alla nascita. Tuttavia, occasionalmente la malattia può associarsi ai sintomi dell'ostruzione addominale (distensione dell'addome, difficoltà di alimentazione, distress respiratorio) quando le cisti diventano particolarmente grandi.
Nel lungo periodo, i pazienti con MCDK monolaterale possono sviluppare ipertensione, proteinuria e insufficienza renale. Questo è dovuto al rischio aumentato di ipertensione e/o proteinuria, espressioni di iperfiltrazione glomerulare o di displasia renale nell'unico rene funzionante. Pertanto, i pazienti con MCDK monolaterale devono essere sottoposti a un follow-up a lungo termine.
Nella via renale controlaterale (quella sana), è aumentata la frequenza di ulteriori anomalie congenite delle vie urinarie (CAKUT), come il reflusso vescico-ureterale e l'ostruzione della giunzione uretero-pelvica (PUJO).
Gestione e Monitoraggio
L'involuzione delle cisti nella MCDK esita in un piccolo rene residuo. Nel 5% dei pazienti con MCDK si osserva un'involuzione prenatale completa, mentre nel 50% l'involuzione completa avviene nella prima decade di vita. Le dimensioni del rene sinistro nel caso di un bambino di 25 mesi, riscontrato in epoca prenatale, possono ridursi a circa 3 cm, con una singola cisti residua di circa 10 mm, mentre il rene destro può presentarsi con dimensioni intorno al 75° percentile, non in franca ipertrofia.
L'esame istologico, quando effettuato, evidenzia cisti circondate da cellule indifferenziate e metaplastiche, occasionalmente con un tessuto renale residuo, funzionante. La renografia con acido dimercaptosuccinico marcato con tecnezio-99m mostra un limitato o assente assorbimento renale, confermando l'assenza di funzionalità del rene multicistico. La diagnosi differenziale si pone con l'ostruzione della giunzione uretero-pelvica (PUJO), nella quale i calici molto dilatati assomigliano a delle cisti.
Fino a poco tempo fa, veniva di solito praticata la nefrectomia, sia per il presunto rischio di ipertensione che per un ipotetico rischio di degenerazione maligna (al momento non supportato da solide evidenze). Oggi, invece, si preferisce non intervenire chirurgicamente e monitorare la MCDK monolaterale con ecografie regolari. Tuttavia, la nefrectomia può essere indicata nei pazienti con ostruzione addominale in presenza di cisti di grosse dimensioni.
Rene policistico: descrizione, cause, sintomi e diagnosi
Altre Malattie Cistiche Renali Ereditarie
Oltre alla MCDK, esistono diverse altre condizioni genetiche che causano lo sviluppo di reni policistici. Queste malattie hanno pattern di trasmissione differenti e possono manifestarsi con gravità variabile, influenzando non solo i reni ma anche altri organi.
Rene Policistico Autosomico Dominante (ADPKD)
Il rene policistico autosomico dominante (ADPKD) è la malattia cistica renale più frequente in assoluto, con un'incidenza di circa 1:1000 nati vivi. Si caratterizza per reni grandi pieni di cisti, che possono generarsi a partire da qualsiasi tratto del tubo renale.
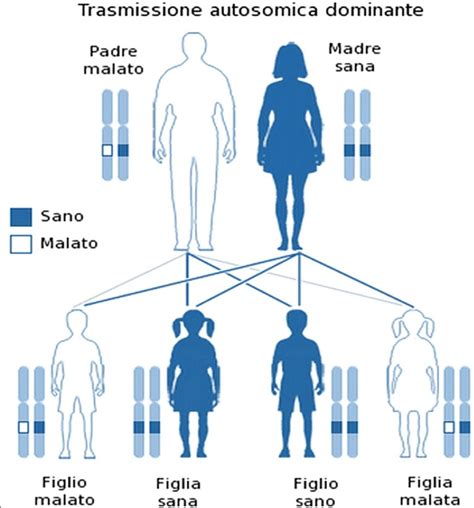
Genetica dell'ADPKD:Questa condizione è dovuta principalmente ad alterazioni dei geni PKD1 (responsabile di circa l'80% dei casi) o PKD2. Più raramente, la malattia è causata da mutazioni in altri geni come GANAB o DNAJB11. Nelle malattie a trasmissione dominante, è sufficiente che una sola copia del gene sia alterata per provocare la malattia; il gene alterato "domina" sull'espressione di quello normale. In questo caso, un genitore affetto ha una probabilità del 50% di trasmettere la mutazione ai propri figli.
Manifestazioni Cliniche dell'ADPKD:Contrariamente a quanto comunemente ritenuto, oggi sappiamo che una percentuale significativa di questi giovani pazienti (20-30%) può presentare ipertensione prima dei 18 anni di vita. In una piccolissima percentuale di casi, la malattia può esprimersi in modo evidente anche nel primo anno di vita, rendendo difficile la distinzione da forme più tipicamente infantili. Possono comparire cisti al fegato, al pancreas e, meno frequentemente, in altri organi, di solito in età adulta, così come aneurismi cerebrali.
Rene Policistico Autosomico Recessivo (ARPKD)
Il rene policistico autosomico recessivo (ARPKD) è una malattia rara che colpisce reni e fegato. I reni sono grandi e la loro struttura è completamente sovvertita dalla presenza di piccole cisti che originano dal dotto collettore. Il fegato è colpito da fibrosi o da dilatazioni diffuse della via biliare.
Genetica dell'ARPKD:Questa condizione è dovuta ad alterazioni del gene PKHD1. Si verifica soltanto nelle persone che hanno ereditato dai propri genitori due copie alterate (mutate) di questo gene. Entrambi i genitori sono "portatori sani", avendo una copia mutata e una normale. Il termine "recessivo" indica che l'alterazione di una sola copia del gene non è sufficiente a provocare la malattia; devono essere entrambe mutate.
Manifestazioni Cliniche dell'ARPKD:La comparsa della malattia può essere molto precoce, talvolta già alla nascita, con ipertensione arteriosa e necessità di dialisi. Nelle forme così gravi, i polmoni possono svilupparsi con difficoltà in gravidanza, manifestandosi con una importante difficoltà respiratoria alla nascita per via di polmoni piccoli. Inoltre, a causa del grande ingombro dei reni in addome, si possono avere difficoltà nell’alimentazione. Nei bambini con malattia renale più grave, durante la gravidanza, si può manifestare l’oligoidramnios, una riduzione della quantità di liquido amniotico dovuta al malfunzionamento renale. Sia il rene che il fegato possono andare incontro a esaurimento funzionale completo, con necessità di dialisi e trapianto. La malattia renale e quella epatica possono associarsi tra loro con diverso grado di gravità e progressione.
Altre Condizioni Renali Cistiche o Associate
Esistono altre patologie che rientrano nel vasto spettro delle malattie cistiche o che presentano anomalie renali congenite con quadri che possono sovrapporsi o essere correlate.
Nefronoftisi
La nefronoftisi, in realtà, non è una singola malattia, ma un gruppo di malattie diverse, accomunate da un quadro renale specifico. Queste patologie possono coinvolgere anche altri organi o apparati. L'ereditarietà è tipicamente autosomica recessiva. Ad oggi, sono noti 21 diversi geni (da NPHP1 a NPHP21) associati a queste malattie renali, che possono presentarsi in forma isolata o essere inserite in sindromi più note, come la sindrome oro-faciale digitale, la sindrome di Jeune, di Joubert, di Meckel-Gruber, di Bardet-Biedl, e altre.
Nella nefronoftisi, i reni sono più spesso di dimensioni normali o solo lievemente ingrandite, fino a ridotte. Altri organi che possono essere coinvolti includono il sistema nervoso centrale, la retina, il fegato, i polmoni e il cuore. Un segno caratteristico nei bambini affetti è il difetto di concentrazione delle urine: producono urine molto chiare, poco concentrate, e per questo possono disidratarsi facilmente se non bevono in modo adeguato.
Sclerosi Tuberosa
La sclerosi tuberosa è una malattia a trasmissione dominante che interessa più organi e apparati, il principale dei quali è il sistema nervoso centrale. Sebbene non sia primariamente una malattia cistica renale, può associarsi alla formazione di cisti renali e altre lesioni renali.
Cisti Renali Isolate
Le cisti renali semplici sono rare nel bambino e solitamente benigne. È importante distinguere le cisti renali semplici da quelle complesse, che potrebbero invece essere indicative di patologie neoplastiche, anche maligne. L'indagine strumentale principale per la diagnosi di queste lesioni è l'ecografia.
Approfondimenti Diagnostici e Genetici
La diagnosi delle malattie cistiche renali si basa su un approccio multimodale che include esami di imaging, valutazioni cliniche e, sempre più spesso, analisi genetiche.
Ruolo dell'Ecografia
L'ecografia, sia in epoca prenatale che post-natale, rappresenta lo strumento diagnostico di prima linea. L'ecografista valuta diversi parametri:
- Presenza della vescica: La sua visualizzazione testimonia l'esistenza di una funzione renale minima (a partire dalla 10ª-11ª settimana di gestazione). La mancata visualizzazione può essere associata a insufficienze placentari gravi, agenesia renale bilaterale, o rene multicistico bilaterale.
- Biometria renale: La valutazione delle dimensioni dei reni. L'ipoplasia renale è diagnosticata se i parametri biometrici sono al di sotto del 10° percentile. Un rene di dimensioni aumentate con regolare ecostruttura può suggerire sindromi come quella di Beckwith-Wiedemann.
- Ecostruttura del parenchima: Un rene aumentato di volume e iperecogeno può indicare rene multicistico, rene policistico, o altre condizioni. La presenza di cisti multiple, non comunicanti, suggerisce displasia multicistica. Reni molto aumentati di dimensioni con parenchima diffusamente iperecogeno sono tipici del rene policistico.
- Dilatazione della pelvi renale: Valutare la dilatazione pielo-caliceale, che può indicare patologie ostruttive delle vie urinarie (es. ostruzione della giunzione uretero-pelvica).
- Localizzazione della patologia: Determinare se la patologia è monolaterale o bilaterale è cruciale per la prognosi e la gestione.
- Presenza di megavescica: Può suggerire patologie come la valvola uretrale posteriore (nei maschi) o la sindrome megavescica-microcolon-ipoperistalsi (nelle femmine).
- Lume vescicale: La presenza di un ureterocele può essere osservata in vescica.
- Genitali: La valutazione dei genitali fetali è importante, specialmente in presenza di altre anomalie, per riconoscere sindromi malformative.
Altre Indagini Diagnostiche
Oltre all'ecografia, altri esami utili includono la scintigrafia renale statica, che valuta la funzionalità renale attraverso l'infusione di un mezzo di contrasto (in caso di MCDK, la funzionalità sarà assente). La cistouretrografia minzionale è un'altra indagine utilizzata per studiare il tratto urinario inferiore.
Diagnosi Genetica
Per molte di queste malattie cistiche renali, è possibile effettuare la diagnosi genetica. Ad esempio, alterazioni nel gene HNF1-beta sono associate a una parte dei casi di displasia cistica e di rene multicistico, e questa condizione si trasmette con modalità autosomica dominante, potendosi associare a diabete, alterazioni epatiche, pancreatiche o degli organi genitali. La diagnosi genetica è fondamentale per confermare la patologia, definire il rischio di ricorrenza nelle famiglie e, in futuro, per lo sviluppo di terapie mirate.
Riflessioni sulla Cura e il Futuro
Le conoscenze sulle malattie cistiche renali fetali e congenite sono in continua evoluzione. Mentre la gestione della MCDK monolaterale si è spostata verso il monitoraggio non invasivo, le forme bilaterali o le sindromi genetiche associate richiedono un approccio multidisciplinare complesso. La ricerca genetica continua a svelare nuovi geni coinvolti e a migliorare la comprensione dei meccanismi patogenetici, aprendo la strada a strategie diagnostiche e terapeutiche sempre più precise e personalizzate per i pazienti e le loro famiglie.
tags: #rene #multicistico #fetale #dimed