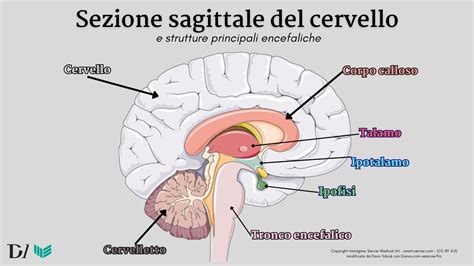
La lissencefalia è uno spettro di rari disturbi congeniti della corteccia cerebrale, caratterizzati da una riduzione della convoluzione corticale, a volte persino dalla sua completa assenza, nota come agiria, o da convoluzioni larghe e appiattite, note come pachigiria. Questa condizione, il cui nome deriva dalle parole greche "lissos" (liscio) e "kephale" (testa) o "encephalos" (cervello), descrive letteralmente un "cervello liscio". In un cervello sano, la corteccia cerebrale è caratterizzata da numerose pieghe (giri) e solchi (solchi), che aumentano la superficie e consentono funzioni cerebrali complesse. Nella lissencefalia, il cervello non riesce a sviluppare queste pieghe, con conseguente superficie liscia. Questa anomalia è il risultato di un difetto nella migrazione neuronale durante il periodo embrionale, che interrompe la normale organizzazione laminare e la formazione delle reti corticali. Tali malformazioni sono significative non solo per il loro impatto sulle funzioni cerebrali, ma anche per le sfide che presentano per le persone colpite e le loro famiglie.
Che Cos'è la Lissencefalia: Una Panoramica Dettagliata
La lissencefalia rappresenta un gruppo di malformazioni della corteccia cerebrale e, secondo le attuali conoscenze, è spesso associata a epilessia, disabilità intellettive e disturbi del movimento. Questi cambiamenti riflettono un difetto fondamentale nella migrazione neuronale, un processo critico che avviene tra la 12ª e la 24ª settimana di gestazione. Durante questa fase, i neuroni appena formati devono spostarsi dalla zona ventricolare, dove nascono, verso la superficie esterna del cervello per formare i sei strati della corteccia cerebrale. Quando questo processo è compromesso, la corteccia risulta abnormemente spessa, con un ordinamento delle circonvoluzioni diminuito o assente, una stratificazione anormale o ridotta della corteccia cerebrale e una diffusa eterotopia neuronale.
La presentazione clinica varia da gravi manifestazioni precoci con marcato ritardo dello sviluppo, ipotonia muscolare, convulsioni e disfunzione della deglutizione, a fenotipi più lievi. Tra questi ultimi, spicca l'eterotopia della banda sottocorticale (SBH), nota anche come "doppia corteccia", in cui è visibile un'ulteriore banda di materia grigia sotto la corteccia. In questa condizione, un'ampia porzione di materia grigia ectopica nella sostanza bianca subcorticale assomiglia a una "doppia corteccia" alla risonanza magnetica. Le diverse cause genetiche spiegano l'ampia diversità fenotipica e le differenze nella prognosi.
Nella Classificazione Internazionale delle Malattie (Decima Revisione, ICD-10), la lissencefalia è codificata sotto "Altre malformazioni congenite dell'encefalo" con il codice Q04.3 "Altre malformazioni riduttive dell'encefalo". Alcuni adattamenti nazionali forniscono una corrispondenza diretta tra il termine "lissencefalia" e termini strettamente correlati come pachigiria e microgiria, il che è importante per la contabilità e il rimborso assicurativo. Nella Classificazione Internazionale delle Malattie (Undicesima Revisione, ICD-11), la lissencefalia è classificata nella sezione "Sindromi con anomalie del sistema nervoso centrale come caratteristica principale" con il gruppo di codici LD20, in particolare "Sindromi con lissencefalia come caratteristica principale" con il codice LD20.1. Condizioni correlate sono riportate anche nella sezione "Disturbi della migrazione neuronale", dove la lissencefalia è esclusa come entità nosologica indipendente, il che ne sottolinea lo status speciale.
Epidemiologia e Incidenza della Lissencefalia
La lissencefalia è una malattia molto rara. Le stime collocano l'incidenza delle forme classiche tra pochi e decine di casi ogni 1.000.000 di nati vivi. Questo dato evidenzia la sua natura eccezionale, rendendola una sfida diagnostica e terapeutica. Tuttavia, il gruppo combinato di malformazioni corticali è significativamente più comune ed è una delle principali cause di epilessia infantile e disabilità intellettiva. Le differenze regionali nelle statistiche di incidenza sono spesso legate alla disponibilità di risonanza magnetica per immagini (RMN) ad alta risoluzione e di diagnostica genetica avanzata, che migliorano il rilevamento di queste condizioni.
Lissencefalia e pachigiria rappresentano una percentuale significativa delle malformazioni corticali, ma sono meno comuni di condizioni come la polimicrogiria e la displasia corticale focale. Tuttavia, nei fenotipi gravi di lissencefalia, la percentuale di epilessia grave e disturbi dello sviluppo è più elevata, il che influisce sulla necessità di cure specialistiche e sull'impatto sul sistema sanitario e sulle famiglie.
L'eterotopia della banda sottocorticale (SBH) è più comunemente diagnosticata nelle ragazze a causa di mutazioni nel gene della doppia corticina legato al sesso (DCX) e può rimanere sottodiagnosticata fino all'adolescenza, quando si manifesta con l'epilessia. Lo sviluppo della risonanza magnetica ad alto campo ha notevolmente aumentato il rilevamento di queste forme più sottili della condizione.
Le stime del carico di malattia includono non solo i costi medici diretti, ma anche l'impatto a lungo termine sulle famiglie e sulla società, dovuto alla necessità di assistenza continua e ai costi indiretti legati alla perdita di produttività e al supporto. La conferma precoce del sottotipo genetico consente una previsione più accurata della storia naturale e del rischio di epilessia e la fornitura di supporto personalizzato, riducendo la necessità di procedure invasive e migliorando la gestione complessiva.
Le Cause della Lissencefalia: Un'Origine Genetica Complessa
Le cause della lissencefalia sono in gran parte genetiche e coinvolgono interruzioni cruciali nella migrazione neuronale. Le sindromi incluse in questa categoria ICD-11 non riguardano solo la morfologia cerebrale, ma si presentano spesso come quadri multisistemici complessi.I geni più comunemente implicati sono quelli associati alle proteine dei microtubuli e al trasporto, come PAFAH1B1, noto come LIS1, e DCX, che codifica per la corticina duale. Alterazioni in questi geni spiegano rispettivamente la lissencefalia classica e l'eterotopia della banda sottocorticale. Mutazioni nel gene LIS1 sono la causa più comune di lissencefalia e sono responsabili della forma autosomica, mentre mutazioni del gene DCX sono responsabili della forma X-linked. Sebbene mutazioni in entrambi i geni possano causare sia lissencefalia sia SBH, la maggior parte dei casi di lissencefalia isolata (ILS, Isolated Lissencephaly Sequence) è dovuta a delezioni o mutazioni del gene LIS1, mentre la maggior parte dei casi di SBH è dovuta a mutazioni del gene DCX. Le delezioni del gene LIS1 causano una forma di lissencefalia più grave nelle regioni posteriori dell’encefalo (gradiente postero-anteriore, p>a), mentre mutazioni di DCX causano una forma di lissencefalia più grave nelle regioni anteriori dell’encefalo (gradiente antero-posteriore, a>p).
Oltre a LIS1 e DCX, sono state descritte alterazioni nei geni TUBA1A, RELN, NDE1, KATNB1, CDK5, ARX e in molti altri. Questi geni regolano l'assemblaggio del citoscheletro, l'orientamento della divisione cellulare, l'attaccamento cellulare e la migrazione direzionale dei neuroni. Le varianti poligeniche e a mosaico spiegano parzialmente la variabilità fenotipica, anche all'interno di una singola famiglia, rendendo il quadro genetico ancora più complesso. Ad esempio, una forma specifica di lissencefalia associata a genitali ambigui nei maschi è dovuta a mutazioni del gene omeobox ARX localizzato sul cromosoma X. Queste mutazioni portano alla sindrome XLAG (Lissencefalia X-linked con agenesia del corpo calloso e genitali ambigui), una grave sindrome malformativa osservata solo nei maschi, caratterizzata da lissencefalia con gradiente P>A, moderato incremento dello spessore corticale, assenza del corpo calloso, microcefalia, epilessia dalla nascita, disfunzioni ipotalamiche e genitali ambigui.
Le cause non genetiche sono meno comuni e possono includere fattori prenatali che interrompono lo sviluppo corticale. Ad esempio, alcune infezioni durante la gravidanza possono contribuire allo sviluppo della lissencefalia, come l'infezione da citomegalovirus (CMV) primaria materna o l'infezione da virus Zika, che è stata collegata a malformazioni cerebrali, inclusa la lissencefalia. Tuttavia, nella maggior parte dei casi, vengono rilevati cambiamenti ereditari o spontanei (de novo). I pannelli di sequenziamento estesi consentono la conferma della causa in una percentuale significativa di pazienti e forniscono una consulenza genetica fondamentale alla famiglia.
In alcuni sottotipi, viene descritto un danno combinato al cervelletto e al tronco encefalico, con un decorso più grave e una compromissione precoce delle funzioni vitali. La comprensione delle basi molecolari consente lo sviluppo di modelli di cura mirati e la previsione delle esigenze di supporto respiratorio, nutrizione e controllo delle crisi epilettiche, migliorando così la gestione clinica.
Neurulazione - Embriologia animata
Meccanismi Patogenetici: Il Difetto della Migrazione Neuronale
Il meccanismo chiave alla base della lissencefalia è l'interruzione della migrazione neuronale diretta dalla zona periventricolare alla corteccia durante il periodo embrionale. Questo processo è finemente regolato e qualsiasi difetto può avere conseguenze devastanti per l'architettura cerebrale. Difetti nelle proteine dei microtubuli, nelle proteine motrici e nelle molecole di segnalazione portano a una migrazione ritardata, a un posizionamento neuronale anomalo e all'interruzione dell'architettura laminare della corteccia.
Il risultato di questa anomalia è una corteccia ispessita, con circonvoluzioni levigate o completamente assenti (agiria) e una stratificazione anomala. I disturbi della migrazione sono accompagnati da cambiamenti secondari nell'organizzazione della rete cerebrale, inclusi percorsi anomali di eccitazione e inibizione, che aumentano la suscettibilità alle crisi epilettiche, una delle manifestazioni cliniche più comuni e difficili da gestire.
L'approccio di rete considera la lissencefalia come un difetto nella formazione di nodi e connessioni che determinano la normale distribuzione dell'attività cerebrale. I meccanismi molecolari influenzano il trasporto di organelli e proteine, la regolazione della polarità cellulare e le interazioni con la matrice extracellulare, tutti elementi cruciali per il corretto posizionamento dei neuroni. Sono stati identificati "gradienti" caratteristici nella gravità del danno per numerosi geni mediante risonanza magnetica, che riflettono differenze regionali nella vulnerabilità corticale. Il rapporto tra agiria e pachigiria, così come il coinvolgimento del cervelletto e del tronco encefalico, sono correlati alla gravità delle manifestazioni cliniche, tra cui difficoltà respiratorie, disturbi della deglutizione e frequenza delle crisi epilettiche. Queste osservazioni vengono utilizzate nella stratificazione clinica e radiologica dei pazienti, consentendo una migliore comprensione della prognosi individuale.
Sintomi e Manifestazioni Cliniche nel Neonato
Il quadro clinico delle sindromi con lissencefalia è generalmente grave e si manifesta precocemente, spesso già nei primi mesi di vita. Le manifestazioni tipiche nell'infanzia includono opistotono, ipotonia muscolare (che può evolvere in ipertonia degli arti), difficoltà di alimentazione e deglutizione (disfagia), e un marcato ritardo nello sviluppo motorio e del linguaggio. Molti bambini sviluppano precocemente un'epilessia generalizzata o focale, che è spesso difficile da trattare. Il sintomo neurologico più comune e difficile da gestire è l'epilessia, che colpisce oltre il 90% dei pazienti. Spesso le crisi iniziano sotto forma di spasmi infantili (Sindrome di West) e possono evolvere in forme di epilessia farmacoresistente. Circa l’80% dei pazienti con lissencefalia classica ha spasmi infantili. Alcuni pazienti sperimentano prevalentemente crisi epilettiche con esordio nei primi mesi di vita, inclusi spasmi e crisi tonico-cloniche generalizzate.
La gravità dei sintomi dipende dall'estensione della lesione e dalle anomalie associate. Nell'eterotopia della banda sottocorticale (SBH), le caratteristiche cliniche sono generalmente meno gravi, ma includono comunque epilessia e deficit cognitivo. In questi casi, l'epilessia può manifestarsi più tardi, durante l'età scolare o l'adolescenza. Le funzioni cognitive sono correlate allo spessore della banda e al grado di pachigiria. L’epilessia è presente in quasi tutti i pazienti con SBH ed è intrattabile nel 65% dei casi.
Oltre alle crisi convulsive, si osserva quasi costantemente un grave ritardo nello sviluppo psicomotorio. Le caratteristiche cognitive e comportamentali includono disabilità intellettiva da moderata a grave, disturbo da deficit di attenzione, tratti autistici e disturbi del sonno. Spesso sono necessari programmi educativi speciali e interventi a lungo termine per supportare lo sviluppo e la qualità della vita di questi bambini.
Nei casi più gravi, si verificano difficoltà respiratorie, apnea e aspirazioni ricorrenti, che possono portare al ricovero ospedaliero e alla necessità di supporto nutrizionale tramite sondino nasogastrico o gastrostomia. Le polmoniti ab ingestis, causate dall'inalazione di cibo o saliva nei polmoni, sono una causa frequente di complicanze. Un'assistenza multidisciplinare tempestiva riduce l'incidenza delle complicanze e migliora significativamente la qualità di vita della famiglia e del bambino.
Classificazione Clinica e Radiologica dei Sottotipi
La lissencefalia è un termine ampio che comprende diverse manifestazioni e sottotipi, la cui classificazione è fondamentale per la prognosi e la gestione clinica. La classificazione clinica e radiologica si basa sulla gravità della levigazione corticale, sull'estensione della lesione e sul coinvolgimento di strutture cerebrali aggiuntive. Si distingue uno spettro che va dall'agiria completa (assenza totale di circonvoluzioni) alla pachigiria regionale (convoluzioni larghe e appiattite), così come l'eterotopia della banda sottocorticale (SBH) come variante del disturbo della migrazione con uno strato aggiuntivo di materia grigia sotto la corteccia.
I sottotipi genici presentano pattern di risonanza magnetica caratteristici. Ad esempio, per le mutazioni nel gene PAFAH1B1 (LIS1), viene spesso descritto un gradiente di espressione dalle regioni occipitale a quella frontale, indicando una maggiore gravità posteriore. In contrasto, per le mutazioni nel gene DCX (doppia corticina), le regioni anteriori e la striscia sottocorticale sono più pronunciate, con un gradiente antero-posteriore. Queste caratteristiche aiutano a suggerire il gene colpevole prima della conferma molecolare.
Storicamente, si distinguono due grandi gruppi di lissencefalia:
- Lissencefalia Classica (Tipo 1): Caratterizzata da una corteccia ispessita che presenta quattro strati, rispetto ai sei normali. Questa è la forma più comune e spesso associata a mutazioni nei geni LIS1 o DCX. Nelle varianti della lissencefalia classica, possono essere presenti anomalie extracorticali, come l'agenesia totale o subtotale del corpo calloso e/o l'ipoplasia cerebellare. Le forme di lissencefalia classica possono essere isolate o associate a sindromi più complesse. Ad esempio, la Sindrome di Miller-Dieker è una condizione causata da delezioni della regione 17p13.3 che comprende LIS1 e geni contigui. I pazienti con questa sindrome presentano lissencefalia classica accompagnata da dismorfismi facciali caratteristici (fronte prominente, leggero ipertelorismo, pieghe dell'epicanto, naso corto e narici anteverse), grave ritardo, epilessia e problemi di nutrizione.
- Lissencefalia con Aspetto Ciottolato (Tipo 2): Questa forma è caratterizzata da una completa disorganizzazione dell'organogenesi cerebrale, con una superficie corticale irregolare e un aspetto "ciottolato" o "zigrinato". Alla microscopia, si osserva la completa disorganizzazione della corteccia e l'assenza di strati distinguibili. La lissencefalia di tipo 2 può essere presente in tre condizioni cliniche principali: le sindromi di Walker-Warburg, MEB (muscolo-occhio-cervello) e Fukuyama. In alcuni di questi pazienti vengono identificate mutazioni del gene POMT1 che interviene sulla O-glicosilazione delle glicoproteine. Queste sindromi sono spesso più gravi e possono includere dilatazione ventricolare, anomalie del cervelletto, dell’occhio e labio-palatoschisi.
Altre forme particolari di lissencefalia includono quelle associate a grave ipoplasia cerebellare, anomalie morfologiche facciali, ritardo dello sviluppo ed epilessia (spesso associate a mutazioni del gene RELN), e quadri di lissencefalia di grado variabile associata a ritardo mentale (associata a mutazioni del gene TUBA1A). Documenti di consenso internazionale propongono di considerare la lissencefalia nel quadro delle malformazioni corticali per standardizzare la terminologia e gli approcci all'esame, facilitando così la ricerca e la pratica clinica.
Complicanze e Prognosi della Lissencefalia
La prognosi per gli individui con lissencefalia varia notevolmente a seconda della gravità della condizione, del sottotipo genetico, dell'estensione della malformazione cerebrale e della presenza di complicanze associate. Generalmente, la prognosi è severa. La sopravvivenza dipende dalla gravità delle crisi e dalla presenza di altre complicanze, tra cui le disfunzioni di deglutizione, l'apnea e la difficoltà nella rimozione delle secrezioni orofaringee. La maggior parte dei soggetti colpiti da lissencefalia grave muore entro i cinque anni di vita, spesso a causa di complicazioni respiratorie.
Le complicanze più comuni sono legate all'epilessia, che è intrattabile in una percentuale significativa di pazienti. Questo include il rischio di lesioni durante le crisi generalizzate e le complicanze respiratorie dovute a stati epilettici prolungati o a disfunzioni neurologiche. Le crisi gravi e lo stato epilettico richiedono cure di emergenza e possono comportare il ricovero in terapia intensiva.
La progressione a lungo termine è accompagnata da difficoltà di alimentazione, malnutrizione, malattia da reflusso gastroesofageo (GERD) e infezioni ricorrenti del tratto respiratorio dovute all'aspirazione. La prevenzione di queste complicanze include il supporto nutrizionale adeguato, la correzione della deglutizione (spesso con l'ausilio di logopedisti e terapisti della deglutizione) e il posizionamento corretto del paziente per ridurre il rischio di aspirazione.
Le disabilità dello sviluppo comportano la necessità di una riabilitazione a lungo termine, dell'uso di dispositivi di comunicazione assistita e di ausili tecnici. Le difficoltà cognitive e comportamentali richiedono percorsi educativi individualizzati e supporto psicosociale, con programmi educativi speciali e interventi a lungo termine. Le famiglie devono affrontare crescenti oneri emotivi ed economici significativi; la consulenza genetica e il supporto della comunità aiutano i pazienti a pianificare il futuro e ad accedere ai programmi di assistenza sociale. L'accesso precoce a centri specializzati migliora il coordinamento delle cure, consentendo un approccio multidisciplinare che coinvolge neurologi, pediatri, genetisti, terapisti e assistenti sociali.
Con un intervento precoce e cure complete, alcuni individui possono raggiungere un certo grado di miglioramento funzionale e una migliore qualità della vita, sebbene le prospettive a lungo termine varino notevolmente. Alcuni individui possono raggiungere un certo livello di indipendenza, mentre altri potrebbero aver bisogno di supporto per tutta la vita.
Quando Ricercare Attenzione Medica Immediata
È fondamentale riconoscere i segnali che richiedono un'immediata attenzione medica per i bambini affetti da lissencefalia o con sospetto di tale condizione, specialmente in età neonatale e nei primi mesi di vita.
I motivi per un'immediata attenzione medica includono:
- Episodi di convulsioni, soprattutto se si verificano nei primi mesi di vita. Qualsiasi convulsione iniziale richiede una valutazione per escludere cause acute e iniziare tempestivamente il trattamento.
- Episodi di arresto respiratorio o apnea.
- Cianosi della pelle (colorazione bluastra), che indica una mancanza di ossigeno.
- Difficoltà di alimentazione o disfagia grave, con conseguente scarso accrescimento o segni di disidratazione.
- Grave letargia o un improvviso cambiamento nel livello di coscienza del bambino.
In presenza di ritardo nello sviluppo motorio o del linguaggio, una forma insolita del cranio (microcefalia o macrocefalia, o asimmetria), nistagmo (movimenti oculari involontari), ipotonia (tono muscolare ridotto) o episodi di spasmi inspiegabili, è essenziale consultare un neurologo pediatrico. Un invio precoce accelera la diagnosi e l'inizio delle terapie di supporto. In questi casi, una risonanza magnetica ed elettroencefalografia saranno probabilmente raccomandate.
Per le donne incinte provenienti da una famiglia con una variante genetica nota associata alla lissencefalia, si raccomanda di sottoporsi a consulenza prenatale per discutere le opzioni per la diagnosi prenatale e la valutazione non invasiva del rischio. Ciò aiuta a prendere decisioni informate all'inizio della gravidanza.
Se la frequenza delle crisi aumenta, si verificano disturbi respiratori durante il sonno (come l'apnea notturna) e frequenti aspirazioni, il piano di trattamento deve essere rivisto. In questi casi, gli interventi nutrizionali e respiratori devono essere discussi e gli obiettivi delle cure palliative devono essere considerati per migliorare la qualità di vita del bambino e della famiglia e ridurre il rischio di situazioni di emergenza.
Diagnosi: Un Percorso Multidisciplinare
La diagnosi precoce della lissencefalia è fondamentale per gestire i sintomi, pianificare interventi appropriati e fornire un supporto completo al bambino e alla sua famiglia. Il percorso diagnostico è multidisciplinare e integra diverse metodologie.
Il primo passo è una valutazione clinica approfondita:
- Una storia dettagliata della gravidanza e del parto, inclusi eventuali eventi avversi o infezioni materne.
- Caratteristiche dello sviluppo precoce del bambino, con particolare attenzione ai ritardi motori, cognitivi o del linguaggio.
- Una descrizione accurata degli episodi convulsivi, se presenti (tipo, frequenza, durata).
- Difficoltà di alimentazione e respirazione.
- Un esame fisico e neurologico completo, che si concentra sul tono muscolare, sulle caratteristiche cranio-facciali (ad esempio, dismorfismi facciali nella sindrome di Miller-Dieker), sui disturbi oculomotori e sui riflessi.
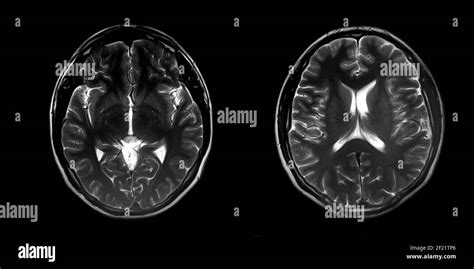
Il secondo passaggio è la neuroimaging, in particolare la risonanza magnetica cerebrale (RMN):
- La RMN cerebrale è l'esame d'elezione e permette di visualizzare chiaramente la superficie liscia del cervello, lo spessore della corteccia (che appare aumentato) e l'eventuale presenza di altre anomalie, come l'ipoplasia del cervelletto o del corpo calloso.
- Si utilizza un protocollo a strati sottili specifico per le malformazioni corticali, con particolare attenzione alle giunzioni cortico-sottocorticali.
- Sono preferiti i sistemi ad alto campo, in quanto migliorano la visualizzazione della corteccia ispessita, delle zone di agiria e pachigiria e dell'eterotopia della banda sottocorticale.
- La diagnosi prenatale può basarsi sull'ecografia, sulla risonanza magnetica fetale o su entrambe. Tuttavia, una proporzione significativa di malformazioni sottili può sfuggire all'imaging prenatale, con il sospetto che può sorgere durante un'ecografia morfologica di routine se si nota una microcefalia o un'assenza dei solchi cerebrali (visibili solitamente dopo la 24ª-26ª settimana).
Il terzo passaggio è costituito dai test genetici molecolari:
- Una volta identificata la malformazione tramite imaging, i test genetici sono fondamentali per determinare la causa esatta e il rischio di ricorrenza per la famiglia.
- Si raccomandano pannelli per malformazioni corticali o sequenziamento dell'intero esoma, con valutazione della patogenicità delle varianti nei geni PAFAH1B1 (LIS1), DCX, TUBA1A, RELN, ARX e altri geni correlati.
- I risultati vengono interpretati in un contesto clinico e radiologico e discussi in una consulenza multidisciplinare con genetisti e neurologi.
Il quarto passaggio è una valutazione dell'epilessia e dello stato somatico generale:
- L'elettroencefalografia (EEG) è essenziale per aiutare a classificare le crisi epilettiche e a pianificare il trattamento anticonvulsivante.
- Gli esami di laboratorio escludono problemi metabolici concomitanti e carenze nutrizionali.
- Test di deglutizione e la valutazione dello stato nutrizionale determinano la necessità di interventi di supporto, come l'alimentazione enterale.
Neurulazione - Embriologia animata
Approcci Terapeutici e Gestione della Lissencefalia
Attualmente non esiste una cura risolutiva per la lissencefalia, poiché non è possibile ripristinare la normale struttura della corteccia cerebrale una volta formata. Gli approcci terapeutici sono di supporto e mirano a gestire i sintomi, prevenire le complicanze e migliorare la qualità di vita dei pazienti e delle loro famiglie.
La gestione dell'epilessia è la priorità clinica e spesso la più difficile. Le crisi epilettiche sono presenti in oltre il 90% dei bambini, con una insorgenza nei primi sei mesi di vita in circa il 75% dei casi, e possono essere farmacoresistenti. È necessario un monitoraggio costante e l'uso di farmaci anticonvulsivanti, spesso in combinazione, per controllare le crisi. La valutazione elettroencefalografica aiuta a guidare la scelta del trattamento.
Il supporto nutrizionale è cruciale. A causa della disfagia (difficoltà di deglutizione) e dei disturbi neurologici, molti bambini faticano ad alimentarsi a sufficienza e presentano scarso accrescimento o malnutrizione. Possono essere necessari interventi come la dieta adeguata, l'alimentazione tramite sondino nasogastrico o la gastrostomia percutanea per prevenire l'aspirazione del cibo e garantire un apporto calorico sufficiente. La gestione del reflusso gastroesofageo è spesso parte integrante di questo supporto.
La fisioterapia e la riabilitazione sono interventi precoci ed essenziali per gestire la spasticità, prevenire contratture articolari, migliorare la postura e, ove possibile, stimolare lo sviluppo motorio. La fisioterapia respiratoria aiuta a evitare i problemi polmonari legati alle aspirazioni e alla debolezza muscolare respiratoria. Sono altresì importanti la terapia occupazionale e la logopedia per affrontare le difficoltà nelle attività quotidiane, nella comunicazione e nella deglutizione.
Per le difficoltà cognitive e comportamentali, possono essere necessari programmi educativi specializzati e un supporto psicosociale. Questo include l'impostazione di un trattamento riabilitativo adeguato e un approccio multidisciplinare che coinvolga neuropsichiatri infantili, psicologi e educatori. L'uso di dispositivi di comunicazione assistita e di ausili tecnici può migliorare l'interazione e l'autonomia del bambino.
I pazienti devono essere sottoposti a un follow-up neurologico, neuropsichiatrico e riabilitativo regolare e a lungo termine. Questo monitoraggio continuo permette di adattare le terapie alle esigenze in evoluzione del bambino e di intervenire tempestivamente in caso di nuove complicanze.
Consulenza Genetica e Prevenzione
Trattandosi in gran parte di una condizione genetica o derivante da eventi casuali durante lo sviluppo embrionale, non esiste una prevenzione primaria assoluta per la lissencefalia nel senso comune del termine. Tuttavia, la consulenza genetica è il passo più importante per i genitori che hanno già un figlio affetto o per famiglie con una storia di lissencefalia.
Identificare la mutazione specifica (ad esempio, nei geni LIS1, DCX, ARX) permette di conoscere il rischio di trasmettere la condizione a futuri figli. Ad esempio, se la mutazione è de novo (nuova nel bambino e non presente nei genitori), il rischio di ricorrenza è molto basso (circa l'1%, dovuto alla possibilità di mosaicismo germinale in uno dei genitori). È corretto effettuare l'analisi della mutazione in entrambi i genitori.
Nel caso in cui venga identificata una mutazione del gene DCX in un maschio con lissencefalia (legata all'X), l'analisi molecolare di DCX deve essere estesa anche alla madre del probando, anche se essa presenta una risonanza magnetica normale. Le donne portatrici di mutazioni DCX possono avere un fenotipo più lieve come l'eterotopia della banda sottocorticale o persino una risonanza magnetica normale. Se la madre è portatrice della mutazione, la trasmetterà con eredità mendeliana. Se la madre non è portatrice, può comunque trasmetterla sotto forma di mosaico germinale. Per tale motivo, è corretto effettuare una diagnosi prenatale a ogni gravidanza di una donna che ha un bambino con caratteristiche di lissencefalia dovuta a mutazioni di DCX.
Analogamente, per le mutazioni del gene ARX, le donne portatrici generalmente hanno un livello cognitivo normale e una risonanza magnetica normale o parziale agenesia del corpo calloso. Tuttavia, sono state descritte alcune donne eterozigoti per mutazioni di ARX che avevano ritardo mentale ed epilessia, sottolineando la variabilità di presentazione.
Le strategie di prevenzione per i fattori non genetici includono misure di salute materna generalmente accettate durante la gravidanza, come evitare infezioni (in particolare quelle da Citomegalovirus e Zika, dove possibile) e l'esposizione a sostanze teratogene. Non esistono fattori di rischio legati allo stile di vita dei genitori (come dieta o attività fisica) che possano causare direttamente la lissencefalia, trattandosi di un errore biologico fondamentale nello sviluppo embrionale. La consulenza genetica offre alle famiglie le informazioni necessarie per prendere decisioni consapevoli riguardo alla pianificazione familiare e alle opzioni di diagnosi prenatale.
Diagnosi Differenziale con Altre Malformazioni Cerebrali
La lissencefalia deve essere distinta da altre malformazioni della corteccia cerebrale e del sistema nervoso centrale che possono presentare sintomi simili o reperti radiologici parzialmente sovrapponibili. La corretta diagnosi differenziale è cruciale per stabilire la prognosi e il piano terapeutico più appropriato.
Tra le principali condizioni da considerare nella diagnosi differenziale vi sono:
1. Polimicrogiria (PMG):La polimicrogiria è una malformazione corticale in cui le circonvoluzioni sono piccole e sovrabbondanti, a differenza della lissencefalia che presenta circonvoluzioni ridotte o assenti. Anch'essa dipende da un difetto di migrazione neuronale. Altri reperti comuni sono la stratificazione corticale elementare o assente nelle regioni colpite, una materia grigia eterotopica, un'ipoplasia o assenza del corpo calloso e del setto pellucido, e le malformazioni del tronco cerebrale e/o del cervelletto. Le anomalie strutturali possono essere diffuse o focali; la zona focale più comunemente colpita è quella attorno alla fessura silviana (bilateralmente o unilateralmente). Il termine sindrome perisilviana è talvolta usato quando i bambini si presentano con segni quali epilessia, debolezza motoria facciale e orale, importanti ritardi del linguaggio e di solito polimicrogiria bilaterale nella regione della fessura silviana. La PMG è frequentemente associata alla schizencefalia, che si caratterizza per la presenza di fessure o fenditure anomale negli emisferi cerebrali. Le manifestazioni cliniche più frequenti sono le convulsioni, la disabilità intellettiva e l'emiplegia o diplegia spastiche. Numerose cause sono state identificate, incluse mutazioni di singoli geni (p. es., di SRPX2) e l'infezione primaria materna da cytomegalovirus.
2. Displasia Corticale Focale (FCD):La displasia corticale focale è una delle malformazioni corticali più comuni e una causa frequente di epilessia farmaco-resistente. Si tratta di un'area localizzata della corteccia cerebrale con architettura anomala, che può presentare neuroni dismorfici e una stratificazione corticale alterata. A differenza della lissencefalia, che è una malformazione diffusa, la FCD è tipicamente circoscritta.
3. Oloprosencefalia:Lo spettro dell'oloprosencefalia si verifica quando il prosencefalo embrionale (che diventa il cervello anteriore) non subisce una completa segmentazione e clivaggio. Questa è una malformazione più grave e strutturalmente distinta dalla lissencefalia. Le oloprosencefalie possono essere causate da mutazioni in un numero di geni (> 14 noti, inclusi quelli del pathway di segnalazione sonic hedgehog). La trisomia 13 e la trisomia 18, così come altre delezioni cromosomiche e duplicazioni, sono state associate all'oloprosencefalia. I tre tipi principali, in ordine decrescente di gravità, sono: alobare (la più grave, solitamente fatale, con fallimento completo del clivaggio e una singola cavità ventricolare), semilobare (clivaggio parziale negli emisferi a livello posteriore ma con una cavità ventricolare unificata comunicante a livello anteriore) e lobare (caratterizzata dall'assenza del setto pellucido, agenesia del corpo calloso, fusione delle corna anteriori dei ventricoli laterali e, possibilmente, fusione del giro del cingolo). Un quarto tipo raro, chiamato variante interemisferica intermedia, è caratterizzato dalla fusione dei lobi frontali e parietali posteriori, nonché possibilmente del talamo, ma con una normale differenziazione emisferica nelle altre sedi. Le anomalie craniofacciali sono presenti nelle oloprosencefalie e il loro grado di intensità di solito rispecchia l'anormalità cerebrale sottostante, includendo anoftalmia o ciclopia, narici malformate o assenti, ipotelorismo, labbro leporino e palatoschisi, e un incisivo centrale. I feti gravemente colpiti possono morire in utero e, dopo la nascita, le manifestazioni comprendono convulsioni, disabilità intellettiva, basso tono muscolare e ritardi motori. Il trattamento è di supporto.
4. Rombencefalosinapsi:La rombencefalosinapsi è una malformazione simile all'oloprosencefalia, ma coinvolge principalmente il cervello posteriore. In questa condizione, vi è fusione degli emisferi cerebellari con parziale o completa assenza del verme (la porzione mediana del cervelletto). Questa malformazione può causare stenosi acqueduttale e idrocefalo. Altre possibili anomalie associate comprendono l'oloprosencefalia dell'encefalo, l'assenza dei bulbi olfattivi, la disgenesia del corpo calloso o del setto pellucido e il VACTERL (vertebral anomalies, anal atresia, cardiac anomalies, tracheoesophageal fistula, renal anomalies, and limb anomalies [anomalie vertebrali, atresia anale, anomalie cardiache, fistola tracheoesofagea, anomalie renali e anomalie degli arti]).
Le malformazioni degli emisferi cerebrali, in generale, possono essere dovute a cause genetiche o acquisite (come infezioni o cause vascolari/metaboliche che interrompono l'apporto di sangue o nutrienti al cervello in sviluppo). Microcefalia o macrocefalia, disabilità motorie da moderate a gravi, disabilità intellettive ed epilessia spesso si verificano con questi difetti, con manifestazioni altamente variabili. La diagnosi precisa, quindi, si basa su una combinazione di reperti clinici, neurologici e, soprattutto, su immagini di risonanza magnetica cerebrale ad alta risoluzione, integrate con test genetici molecolari specifici.
tags: #lissazione #anche #lattante