La gravidanza rappresenta un periodo di attesa e di profondo cambiamento, durante il quale la salute e lo sviluppo del nascituro sono al centro dell'attenzione. In questo contesto, il monitoraggio accurato delle condizioni fetali è di fondamentale importanza. Tra le diverse condizioni che possono influenzare l'andamento di una gravidanza, la sindrome di Down e le alterazioni del liquido amniotico assumono un rilievo significativo, sia per la loro frequenza che per le implicazioni cliniche e diagnostiche. Questo articolo si propone di esplorare la natura della sindrome di Down, le funzioni e le anomalie del liquido amniotico, e come queste due aree si intersecano nel complesso percorso della diagnosi e gestione prenatale, fornendo un quadro dettagliato per comprendere meglio queste importanti tematiche.
La Sindrome di Down: Un Quadro Genetico Approfondito
La sindrome di Down, conosciuta anche come trisomia 21, è una condizione genetica che si verifica a causa della presenza di un cromosoma 21 in più nelle cellule del corpo, ossia tre copie invece delle normali due. Questo cromosoma extra altera lo sviluppo fisico e cognitivo, determinando una disabilità intellettiva di gravità variabile e altri problemi di salute. In Italia, la sindrome di Down è riconosciuta nell'elenco delle patologie croniche e invalidanti, esente dal costo del ticket.
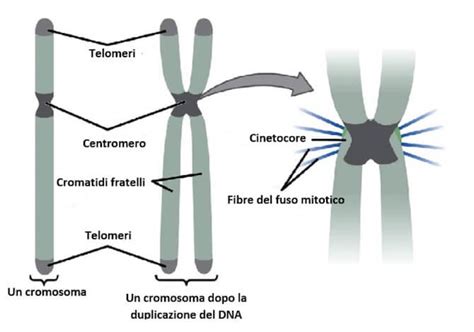
Normalmente, le cellule del corpo umano contengono 46 cromosomi, con un bambino che eredita 23 cromosomi dalla madre e 23 cromosomi dal padre. Nelle persone con sindrome di Down, tutte le cellule, o alcune di esse, contengono 47 cromosomi, poiché vi è una copia in più del cromosoma 21. La presenza di un cromosoma supplementare, ossia tre copie dello stesso cromosoma (invece delle normali due), viene definita trisomia. La trisomia più comune nei neonati è la trisomia 21, che rappresenta circa il 95% dei casi di sindrome di Down. Questa copia extra del cromosoma 21 è solitamente dovuta a un errore nella divisione cellulare dello spermatozoo o della cellula uovo prima del concepimento, un fenomeno chiamato “non disgiunzione”.
Esistono altre forme della sindrome, sebbene meno frequenti. La sindrome di Down da traslocazione rappresenta circa il 4% dei casi e si verifica quando nel cariotipo, ovvero nell'insieme dei cromosomi, di uno dei genitori è presente una parte del cromosoma 21 in più "attaccata" a un altro cromosoma, spesso il 14, 15 o, più raramente, il 21. I genitori con questa traslocazione hanno un'alta probabilità di generare uno spermatozoo o una cellula uovo con una frazione del cromosoma 21 soprannumeraria. Quando non si ha né perdita di materiale genetico né aggiunta, come in questo caso, si parla di traslocazione bilanciata, e un genitore può esserne portatore, con la possibilità di trasmetterla. Il mosaicismo, invece, rappresenta circa l'1-2% dei casi di sindrome di Down. Questa condizione si verifica dopo il concepimento e di conseguenza la trisomia 21 non è presente in tutte le cellule dell'individuo. In questo tipo di sindrome di Down è presente una miscela di due tipi di cellule: alcune contengono i soliti 46 cromosomi e alcune contengono 47 cromosomi, con la prevalenza dell’uno o dell’altro tipo di cellule che fa la differenza nella forma più o meno lieve della sindrome.
Incidenza e Fattori di Rischio
In ogni gravidanza esiste una probabilità, seppur bassa, di avere un bambino con sindrome di Down. L'incidenza di nati vivi con la sindrome a livello mondiale è di circa uno su 1.000, mentre in Italia si stima che circa un bambino ogni 1.200 nati abbia la sindrome, corrispondente a circa 500 nascite all’anno per un totale di 38.000 persone nel Paese. La sindrome di Down è presente in tutte le etnie, senza alcuna distinzione, e interessa sia maschi che femmine.
Attualmente, non ci sono evidenze che dimostrino che la sindrome possa essere causata da particolari fattori o attività svolte prima o durante il periodo della gravidanza, sebbene alcune persone presentino un rischio maggiore. Tra i principali fattori di rischio, ad esempio, è riconosciuta l'avanzata età della madre al momento del concepimento. Il rischio di una coppia di avere un bambino con un cromosoma supplementare aumenta progressivamente con l'aumentare dell'età della madre, in particolare dai 35 anni in su. Tuttavia, dato che la maggior parte dei bambini nasce da donne giovani, solo circa il 20% dei neonati con sindrome di Down nasce da madri sopra i 35 anni. Circa l'80% dei nati con sindrome di Down riguarda gravidanze in donne di età inferiore ai 35 anni, poiché in questa fascia di età si concepiscono e nascono più bambini.
Nonostante non sia stata molto approfondita, esiste anche un'influenza, seppur inferiore (5-6%), dell'età paterna sulla possibilità di avere un bambino con sindrome di Down. La probabilità aumenta anche nel caso in cui la trisomia 21 sia già stata accertata in una precedente gravidanza; dopo una gravidanza con trisomia 21 "libera", il rischio in una successiva gravidanza è circa l'1% oltre al rischio legato all'età materna. Se la trisomia 21 è dovuta a traslocazione Robertsoniana, è indicato il cariotipo dei genitori. Con genitori normali, il rischio residuo è in genere ~2-3%; se un genitore è portatore, il rischio dipende dal sesso del portatore e dal cromosoma coinvolto: madre portatrice 10-15%, padre portatore ~1-3%. Anche lo status socio-economico può influire, probabilmente a causa di un accesso più complicato ai programmi sanitari di screening e una minore consapevolezza verso uno stile di vita più salutare.
Caratteristiche e Comorbilità della Sindrome di Down
Bambini e bambine con sindrome di Down hanno difficoltà di sviluppo di grado variabile. Nei primi anni di vita è possibile riscontrare un ritardo nel raggiungimento delle tappe dello sviluppo psicomotorio. Spesso i bambini con sindrome di Down sono di bassa statura e presentano un rischio aumentato di sviluppare obesità. Sviluppo fisico e mentale sono caratterizzati da ritardo. Il cranio è di norma piccolo e potrebbe presentare un appiattimento a livello occipitale; le fontanelle, nei neonati, sono larghe e si chiudono in ritardo rispetto alla norma. I neonati con sindrome di Down tendono a essere calmi e passivi e piangono meno del normale. Tendono ad avere una testa piccola e un volto largo e piatto con naso corto. Nella parte posteriore del collo può essere presente un eccesso di cute (pieghe nucali).
Le caratteristiche fisiche tipiche dei soggetti con sindrome di Down possono includere ipotonia, fessura palpebrale obliqua in alto, occhi a mandorla, piega palmare singola, statura ridotta. Gli occhi hanno un tipico taglio inclinato verso l’alto e la pelle della palpebra superiore copre l’angolo interno dell’occhio (plica epicantale). Talvolta, la lingua è grande; spesso, la lingua più grande e il tono muscolare del volto ridotto fanno sì che i bambini tengano la bocca aperta. Le orecchie sono piccole e arrotondate e hanno un impianto basso nella testa. Di solito le mani sono corte e larghe, con un’unica piega palmare. Le dita sono corte e il dito mignolo, che spesso ha due falangi invece di tre, è curvato verso l’interno. Tuttavia, ogni persona è unica e può presentare tali caratteristiche in misura variabile o non presentarle affatto; alcuni neonati non presentano tratti del viso caratteristici evidenti alla nascita e li sviluppano invece durante la prima infanzia.
Le persone con sindrome di Down possono presentare alcuni disturbi con una frequenza superiore alla norma. Tra questi: problemi cardiaci, intestinali, difficoltà nella vista e nell'udito e un aumento del rischio di infezioni. Le cardiopatie congenite riguardano circa il 50% delle persone con sindrome di Down, essendo i più comuni difetto del setto interventricolare e difetto del setto atrioventricolare. Questi difetti possono essere gravi al punto da richiedere controlli periodici e interventi chirurgici già nella prima infanzia. I disturbi intestinali variano dalla costipazione, alla diarrea, all'indigestione sino a malattie più gravi come la stenosi duodenale. Alcuni bambini sviluppano la celiachia e la malattia da reflusso gastroesofageo.
Circa l'80% delle persone con sindrome di Down, di qualsiasi età, può presentare un difetto dell'udito più o meno grave; nel primo anno di vita può comparire una caratteristica otite sierosa che può persistere nel tempo. La prevenzione dei problemi uditivi è molto importante per evitare che si aggiungano difficoltà allo sviluppo di una buona capacità di comunicazione e di socializzazione. Anche i problemi della vista sono abbastanza frequenti; circa il 60% delle persone con sindrome di Down presenta problemi oculari come cataratte, glaucoma e strabismo.
Rispetto alla popolazione generale, le persone con sindrome di Down hanno un rischio più elevato di sviluppare alterazioni a carico della tiroide, una ghiandola che produce ormoni implicati nello sviluppo fisico e mentale dei bambini. La possibilità che si verifichi un ipotiroidismo è più frequente e può essere dovuta all'assenza della tiroide alla nascita (ipotiroidismo congenito) o all'attacco della tiroide da parte del sistema immunitario (ipotiroidismo autoimmune). Le persone con sindrome di Down hanno una maggiore sensibilità alle infezioni perché il loro sistema immunitario non si sviluppa in modo del tutto adeguato. Presentano anche un rischio più elevato di sviluppare leucemia e un rischio molto più elevato di sviluppare apnea ostruttiva del sonno. Il quoziente intellettivo ha ampia variabilità intra-sindromica: raramente è normale e generalmente è compreso tra 50 e 80. Il linguaggio è la funzione strettamente legata allo sviluppo cognitivo che appare più compromessa rispetto all'organizzazione delle altre abilità superiori e al livello intellettivo.
Prognosi e Qualità della Vita
La sindrome di Down ha una prognosi migliore rispetto ad altri disturbi causati da un cromosoma supplementare, come la trisomia 18 o la trisomia 13. Grazie soprattutto al miglioramento della possibilità di trattare le complicanze, l'aspettativa di vita media nei Paesi sviluppati è in aumento, superando oggi i 55-60 anni (negli anni Sessanta era di appena 15). Il processo di invecchiamento può essere accelerato. Sintomi di demenza, quali perdita di memoria, ulteriore peggioramento delle capacità intellettive e cambiamenti della personalità possono svilupparsi in giovane età, in genere prima dei 40 anni. La maggior parte dei decessi dei soggetti con sindrome di Down è dovuta a malattie cardiache, infezioni e leucemie. Non esiste una cura per la sindrome di Down, ma alcuni sintomi e problemi specifici possono essere trattati.
Che cos'è la sindrome di Down - Intervista alla dott.ssa Valentini
Il Liquido Amniotico: Funzione, Dinamiche e Alterazioni
Il liquido amniotico è un liquido chiaro contenuto nel sacco amniotico che circonda e protegge il bambino durante la gravidanza. Inizialmente, il liquido è costituito essenzialmente da acqua. Verso la 20ª settimana di gestazione, l’urina del feto ne diventa la sostanza principale. Il feto respira e inghiotte il liquido amniotico che favorisce la nutrizione, la maturazione dei polmoni, la crescita del feto ed il mantenimento di una temperatura stabile. Il volume del liquido amniotico aumenta durante la gravidanza, raggiungendo il livello massimo all’incirca alla 34ª settimana. La sua quantità diminuisce poi a un tasso del 25% a settimana dopo la 34-36 settimana di gestazione. La quantità di riduzione potrebbe essere di 150 millilitri a settimana durante la 38ª-43ª settimana. Dal momento che un volume anormale di liquido amniotico è associato a una varietà di complicazioni, il monitoraggio del liquido amniotico fatto insieme al profilo biofisico e il test che misura i battiti cardiaci, la respirazione, i movimenti e il tono muscolare contribuisce a stabilire il benessere del feto.
Oligoidramnio: Cause, Diagnosi e Complicanze
Quando il volume di liquido amniotico diminuisce o diventa inadeguato, insorge la condizione di oligoidramnio o oligoidramnios. L’oligoidramnios colpisce dal 4% all’8% delle gravidanze e deve essere necessariamente diagnosticato e gestito in modo tempestivo per prevenire lesioni al bambino.
Cause dell'Oligoidramnio
Il volume del liquido amniotico è determinato dalla quantità di liquido che fluisce nel e dal sacco amniotico. La rottura delle membrane è la causa più comune di oligoidramnio. Dal momento che il liquido amniotico è costituito principalmente da urina del feto durante la seconda metà della gravidanza, l’oligoidramnio può essere causato anche dall’assenza di produzione di urina da parte del feto o da un blocco presente nel tratto urinario del feto. L’oligoidramnio può essere una condizione cronica o acuta. La condizione acuta si presenta nei casi di ipossia fetale e può essere causata da una gestosi (o preeclampsia) molto grave. L’oligoidramnio è raro nel primo trimestre e le cause di questa condizione nell’arco di questo periodo della gravidanza rimangono tuttora oscure. Molte donne che sviluppano oligoidramnios non presentano fattori di rischio identificabili.
Diagnosi di Oligoidramnio
L’oligoidramnio viene generalmente diagnosticato da una visita medica, un’analisi della storia materna ed un’ecografia. In alcuni casi l’ecografia rivela le cause dell’oligoidramnio, come ad esempio una gravidanza gemellare. Si può sospettare la condizione inizialmente quando le dimensioni dell’utero sono ridotte rispetto all’aspettativa per l’età gestazionale. I medici effettuano la diagnosi di oligoidramnio tramite ultrasuoni, ottenendo una misura chiamata indice del liquido amniotico (AFI). Questo indice viene calcolato misurando la profondità del liquido in quattro sezioni dell’utero che poi vengono sommate. In prossimità del termine della gravidanza un indice normale di fluido amniotico è fra gli 8 e 18 cm (a volte viene considerato normale anche un indice fra i 5 e i 15 cm). Fra la 20ª e la 35ª settimana di gestazione viene considerato normale un indice di 14 cm. Se l’indice è minore di 5 cm fra la 32ª e la 36ª settimana, la donna ha l’oligoidramnio, anche se alcuni medici considerano la presenza di questa condizione con un indice minore di 8 cm. Tipicamente l’oligoidramnio viene diagnosticato quando l’indice è al di sotto dei 5 cm, mentre un indice di 5-8 cm viene considerato borderline. Fra la 36ª e la 42ª settimana un indice normale è di 12,9 cm con una variazione di 4,6 cm. Dato che il volume del liquido amniotico dipende dall’età gestazionale, l’oligoidramnio può anche essere definito come un indice di liquido amniotico minore del 5º percentile di un indice normale proprio dell’età gestazionale.
Rischi e Conseguenze
L’oligoidramnios è una condizione molto grave associata al ritardo della crescita del feto. Può anche causare la compressione del cordone ombelicale, l’encefalopatia ipossico-ischemica e altre lesioni gravi al bambino, inclusa la morte. Le complicazioni durante il secondo e terzo trimestre sono spesso dovute alla compressione del cordone ombelicale, all’insufficienza utero-placentare e all’aspirazione di meconio. L’insufficienza utero-placentare e la compressione del cordone ombelicale possono causare anomalie nel battito fetale che richiedono un parto cesareo e danno un basso punteggio Agpar. A causa dell’alto rischio di un esito negativo, queste gravidanze vanno necessariamente tenute sotto stretto controllo ad ogni visita neonatale per verificare la presenza di condizioni acute e a lungo termine, ed è essenziale che la frequenza del battito cardiaco sia monitorata continuamente.
Gestione dell'Oligoidramnio
Non esiste una terapia a lungo termine per gestire l’oligoidramnio. La diagnosi di liquido amniotico ridotto durante il primo trimestre è quasi assente, dal momento che in genere il feto abortisce spontaneamente. Durante questo periodo la gestione e la prognosi dell’oligoidramnio dipende dalla causa e gravità della condizione. In situazioni limite di volume ridotto del liquido amniotico, in genere, si verifica una prognosi positiva. Quando l’oligoidramnio è presente nel secondo trimestre spesso si verifica la morte del feto o del neonato. Il parto prematuro spontaneo avviene nel 50% dei casi.
Se diagnosticato tempestivamente, in molti casi si riesce a gestire con ricovero ospedaliero, monitoraggio del feto, idratazione per via orale o endovenosa e parto immediato nel caso in cui il monitoraggio del battito fetale mostri segni di anomalie. La terapia in genere consiste nel somministrare liquidi per via orale alla madre affinché si reidrati o nell’amniofusione se non si riesce a visualizzare bene il feto. Frequenti ecografie devono essere eseguite per tenere sotto controllo il livello di liquido amniotico, la crescita, lo sviluppo e il benessere del feto. Le complicazioni specifiche della gravidanza associate all’oligoidramnio devono essere gestite in modo appropriato.
Alle donne con gravidanze ad alto rischio, che presentano fattori di rischio per l’oligoidramnio, va effettuato un esame dell’indice del liquido amniotico una volta a settimana se l’età gestazionale è al di sotto delle 41 settimane. Se l’indice è 5-8 cm con un’età gestazionale al di sotto delle 41 settimane di gravidanza, la misurazione va effettuata due volte a settimana per il rischio di avere un indice al di sotto di 4 cm nell’arco di 4 giorni. A tutte le donne che hanno terminato la 41ª settimana di gravidanza va effettuata una misurazione dell’indice due volte a settimana e la frequenza dell’ecografia dipende dalle circostanze cliniche di ogni donna. Quando il liquido amniotico risulta ridotto in un lasso breve di tempo, si dovrebbe considerare l’opportunità di effettuare esami più frequenti e di ricovero in ospedale con monitoraggio fetale costante. Il consenso informato va comunque ottenuto.
Quando la causa dell’oligoidramnio è sconosciuta si indica il monitoraggio costante e non è necessario confermare tramite ecografia la maturità dei polmoni se il parto avviene dopo la 36ª settimana. Il parto indotto incrementa il rischio di dover sottoporre la madre ad un parto cesareo e alle complicazioni associate ad esso. In alternativa si può seguire la madre con frequenti monitoraggi fetali e profilo biofisico fino al termine della gestazione. Se in una gravidanza post-termine è presente l’oligoidramnio si procede subito al parto, al di là del punteggio dal profilo biofisico, e la sorveglianza costante del neonato è fondamentale. L’oligoidramnio in gravidanze post-termine è associata ad un’incidenza maggiore di presenza di meconio nel liquido amniotico e un rischio maggiore di parto cesareo. Durante il travaglio bisogna monitorare continuamente il battito cardiaco.
Aspetti Medico-Legali nella Gestione dell'Oligoidramnio
L’oligoidramnio può avere effetti devastanti. Pertanto è essenziale che il team medico segua le linee guida per la cura, soprattutto se sono presenti i fattori di rischio per la gravidanza. È fondamentale che i medici tengano sotto stretto controllo tramite analisi e monitoraggi frequenti sia la madre che il bambino per prevenire o minimizzare le complicazioni associate all’oligoidramnio. Il medico dovrebbe essere pronto a ricoverare la madre in ospedale per il monitoraggio costante del feto e per un possibile parto cesareo o comunque un parto al più presto possibile. È essenziale che il medico curi tutte le condizioni che possano causare l’oligoidramnio, che sia in grado di diagnosticarlo accuratamente e che possa monitorare attentamente il liquido amniotico mentre scende di livello. Quando l’oligoidramnio non viene gestito in modo appropriato e i fattori di rischio non vengono diagnosticati o curati si verifica un caso di negligenza medica. Quando la madre e il bambino non vengono monitorati con attenzione e non si seguono gli standard per gestire la condizione, si è in presenza di un atto di negligenza.
Polidramnios: La Condizione Opposta
Il polidramnios, che si verifica nell’1% delle gravidanze, è la condizione opposta all’oligoidramnios, in cui vi è un eccesso di liquido amniotico. Se diagnosticata in tempo, è facile da gestire. Il liquido amniotico è prodotto nei reni del feto ed è espulso quando il bambino urina mentre è in utero. Il bambino poi ingoia una certa quantità del liquido. Le cause del polidramnios possono includere la sindrome da trasfusione da gemello a gemello (TTTS), dove in alcune gravidanze gemellari, il sangue di un gemello va all'altro, o patologie del cuore fetale, che potrebbero includere un difetto congenito o aritmia fetale, ecc. Il polidramnios può essere diagnosticato per mezzo dell’ecografia. I casi lievi di polidramnios possono non richiedere un trattamento, ma i medici devono sottoporre i loro pazienti a controlli extra prenatali per monitorare la condizione e assicurarsi che non diventi grave. I trattamenti per forme più severe di polidramnios a volte si concentrano nell’affrontare le cause sottostanti. L'amnioriduzione comporta l’uso di un ago lungo per drenare il liquido amniotico in eccesso dall’utero. Possono essere somministrati anche farmaci per ridurre la quantità di urina fetale, ma questo può mettere a rischio la salute del feto, quindi i medici devono fare attenzione nel determinare se questa sia la strada giusta.
Diagnosi Prenatale della Sindrome di Down: Tra Screening e Metodiche Invasive
Il periodo prenatale è di fondamentale importanza per la preparazione della coppia alla vita futura del bambino che dovrà nascere. Attualmente esistono diversi tipi di test di screening prenatale che possono essere effettuati a partire dal primo trimestre di gravidanza e hanno lo scopo di fornire una stima individuale del rischio di trisomia 21.
Test di Screening Non Invasivi
Lo screening e la diagnosi prenatale devono essere offerti a tutte le persone in gravidanza con informazioni chiare su benefici, limiti e alternative. Il consenso informato e la decisione condivisa sono pilastri fondamentali.
Nel primo trimestre, tra l'undicesima e la quattordicesima settimana di gravidanza, può essere eseguito il test combinato. Questo test include un esame del sangue materno e la misura ecografica della translucenza nucale (spessore di uno spazio compreso tra la cute e la colonna vertebrale dietro la nuca del feto), e consente di stimare il rischio della presenza della trisomia 21 in epoca molto precoce. Tra 11+0 e 13+6 settimane, quando la CRL è compresa fra 45 e 84 mm, si esegue l’ecografia con misurazione della translucenza nucale (NT) e la valutazione di altri marker (osso nasale, flusso nel dotto venoso, rigurgito tricuspide ove previsto dai protocolli), abbinata ai marcatori biochimici materni PAPP-A e hCG libera. Il "test combinato" offre una buona performance di screening per trisomia 21 e altre aneuploidie, con valori riportati in letteratura attorno all’82-90% di rilevazione a falsi positivi ~3-5% in contesti controllati. A questo test si associa un’ecografia fra la 11ª e la 13ª settimana di gravidanza, che valuta lo spessore di uno spazio liquido che si trova in corrispondenza della nuca fetale, che pure consente di individuare circa il 75% dei casi di sindrome di Down.
Il NIPT Test (non invasive prenatal test), o screening prenatale non invasivo (NIPS) o analisi del DNA fetale libero circolante, ricerca il DNA fetale nel sangue materno ed è disponibile a partire dalla decima settimana di gestazione. Per la trisomia 21 la sensibilità e specificità sono molto elevate (≈ >99% e >99% rispettivamente in numerosi studi), mentre il valore predittivo positivo varia con il rischio pre-test (età materna, storia, esito di altri screening). Si tratta di un esame molto sofisticato con una sensibilità e una specificità che si avvicinerebbero al 100%. In Italia fino ad oggi è stato poco utilizzato in quanto molto costoso e non dispensato dal Servizio Sanitario Nazionale, tranne in alcune regioni dove è stato inserito nei livelli essenziali di assistenza (Emilia Romagna, Valle d’Aosta). Il cfDNA resta uno screening: un risultato “ad alto rischio” va confermato con test diagnostico invasivo prima di decisioni cliniche importanti. Fattori che riducono la fetal fraction includono BMI elevato, prelievo troppo precoce, patologie autoimmuni e, in alcuni studi, terapia anticoagulante (eparina), che possono aumentare il tasso di risultati indeterminati. Alcuni test cfDNA offrono l’analisi “genome-wide” di rare autosomal trisomies e microdelezioni.
L’ecografia morfologica tra 18 e 22 settimane è raccomandata per tutte le gravidanze. Il riscontro di soft marker isolati (p. es. l'aumento dell'NT e l'assenza/ipoplasia dell'osso nasale nel primo trimestre) hanno valore aggiunto limitato se lo screening è negativo; l'attenzione si concentra su malformazioni maggiori. La possibilità di individuare un’anomalia dipende dalle dimensioni e dalla posizione del feto e dell’utero; dalla quantità di liquido amniotico e dallo spessore della parete addominale materna; tutti aspetti che possono sfuggire all’esame ecografico e che attribuiscono allo stesso poco valore diagnostico.
Il Tritest, invece, consta di un prelievo di sangue che la gestante può effettuare tra la 15ª e la 17ª settimana per valutare e dosare la presenza di tre sostanze prodotte in parte dalla placenta e in parte dal fegato del feto: l’alfafetoproteina, l’estriolo non coniugato e la beta-gonadotropina corionica. Il Bitest è sovrapponibile al Tritest, anch'esso una stima di rischio non diagnostica, con il vantaggio di poter essere eseguita più precocemente e con esattezza tra l'11ª e la 14ª settimana di amenorrea. I risultati dei test di screening sopracitati danno comunque solo risposte di tipo probabilistico e non hanno nessun valore diagnostico certo; tuttavia, permettono di identificare donne che potrebbero decidere di sottoporsi ad un esame diagnostico invasivo, avendo un rischio più elevato di dare alla luce un bambino Down.
Metodiche Diagnostiche Invasive
Il rischio di partorire un bambino affetto da sindrome di Down può essere valutato prima della nascita tramite numerosi test diagnostici invasivi, che consentono diagnosi citogenetica o genomica del feto. In presenza di malformazioni fetali all’ecografia, il microarray è raccomandato perché aumenta il tasso diagnostico rispetto al cariotipo.
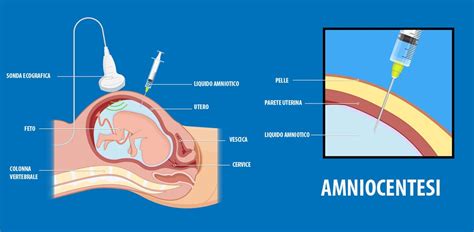
La villocentesi si esegue in genere tra 10 e 13 settimane e campiona i villi coriali placentari; consiste nella analisi di una piccola quantità di tessuto placentare, ovvero un prelievo di cellule da cui si svilupperà la placenta. L’amniocentesi si effettua generalmente intorno alla 15ª settimana di gestazione (≥15 settimane) e campiona liquido amniotico tramite un prelievo, con un sottile ago inserito nella parete addominale, di circa 20 ml di liquido amniotico che possiede cellule di sfaldamento della cute del feto su cui viene effettuata l’analisi cromosomica.
Un altro tipo di test è la cordocentesi, effettuata dalla 18ª settimana in poi; consiste nel prelevare una piccola quantità di sangue fetale dai vasi del cordone ombelicale.
Rischi e Indicazioni per le Metodiche Invasive
Queste metodiche, per quanto utili e mostrando un alto grado di attendibilità, comportano un rischio di perdita precoce abortiva dell’1% per villocentesi e amniocentesi, e pari al 2% per la cordocentesi. Per questo motivo vengono effettuate solo quando è necessario, cioè nei casi di donne considerate a rischio (per esempio donne con un’età superiore ai 35-37 anni) o con un precedente figlio Down. In questo caso, infatti, la probabilità di avere una seconda gravidanza con la stessa anomalia è dell’1%, a meno che il primo feto non abbia una sindrome da traslocazione, nel qual caso la probabilità è maggiore. È più probabile che la fetal fraction sia bassa in caso di gravidanza gemellare quando si usa il cfDNA.
La Celocentesi: Una Nuova Prospettiva
La celocentesi è una innovativa procedura di diagnosi prenatale sviluppata dalla Fondazione Cutino che potrebbe a breve consentire di diagnosticare la sindrome di Down già dal secondo mese di gravidanza. Lo studio sulla celocentesi, che si è concluso nel 2010 ed è stato svolto in 3 anni su 111 gravidanze a rischio talassemia, ha già portato a 232 diagnosi precoci per Talassemia. La celocentesi fornisce risultati certi al 100% già dal secondo mese di gravidanza, consentendo di ricorrere all’interruzione volontaria di gravidanza e non all’aborto terapeutico, con un beneficio per la donna sia fisico che emotivo. Inoltre, la diagnosi così precoce apre la strada a interventi terapeutici in utero. Si aprono, dunque, nuovi orizzonti diagnostici per la sindrome di Down, esplorando la possibilità di eseguire lo studio del cariotipo fetale su DNA estratto da cellule del liquido celomatico.
Il Percorso Diagnostico: Dalla Stima del Rischio alla Conferma
Indipendentemente dall’età, a tutte le gestanti è consigliato l’esame per la sindrome di Down prima della 20ª settimana di gestazione. L'offerta del NIPT tramite SSN varia per Regione in termini di universalità, criteri di accesso, fasce di rischio o progetti pilota. In alternativa è disponibile in intramoenia/privato. Lo screening negativo non azzera il rischio, e un cfDNA “alto rischio” non è una diagnosi, ma richiede conferma. Il cfDNA riflette il genoma placentare, quindi mosaicismo placentare confinato, mosaicismo materno o raramente condizioni materne (p. es. neoplasie) possono spiegare risultati cfDNA discordanti con il cariotipo fetale.
Gestione e Supporto Post-Natale per la Sindrome di Down
Alla nascita, la diagnosi di sindrome di Down viene fatta sulla base delle caratteristiche fisiche del neonato. Dopo la nascita, il tipico aspetto fisico di un neonato affetto da sindrome di Down suggerisce la diagnosi, che viene confermata con un esame del sangue del neonato, per l'individuazione di una terza copia del cromosoma 21, di solito mediante l'analisi di un campione di sangue. Viene eseguito anche il test della conferma del cariotipo, per il numero dei cromosomi, e viene valutata la possibile coesistenza di altre patologie genetiche o congenite, non diagnosticabili nel periodo prenatale.
Controlli Clinici e Prevenzione delle Comorbilità
Dopo aver formulato la diagnosi, i medici eseguono altri esami per individuare le anomalie associate alla sindrome di Down. Il trattamento delle anomalie rilevate può spesso prevenire il peggioramento delle condizioni di salute. Le persone con sindrome di Down hanno bisogno di sottoporsi a regolari controlli clinici per la prevenzione o diagnosi in fase precoce delle malattie associate alla loro condizione. Questi esami vengono eseguiti a determinati intervalli e includono ecografia del cuore, esami del sangue che includono la funzione tiroidea, esame della vista, test dell’udito. Le misurazioni di altezza, peso e circonferenza cranica vengono registrate a ogni bilancio di salute utilizzando una tabella della crescita creata specificamente per i bambini con sindrome di Down, e valutazione dell’apnea ostruttiva del sonno. I bambini con dolore al collo o ai nervi, debolezza o altri sintomi neurologici devono essere sottoposti a radiografia delle articolazioni del collo per rilevare eventuale instabilità. Anche i bambini e gli adulti che desiderano partecipare alle paralimpiadi o ad altri eventi sportivi possono avere bisogno di un esame radiografico delle articolazioni del collo. Gli adolescenti e gli adulti con sindrome di Down devono essere sottoposti a screening a determinati intervalli per il diabete, la tiroide ipoattiva (ipotiroidismo) e la malattia di Alzheimer.
Approccio Multidisciplinare e Interventi di Supporto
Non esistono cure per la sindrome di Down, tuttavia alcuni sintomi e problemi specifici causati dalla stessa possono essere trattati. I medici possono riparare chirurgicamente alcuni difetti cardiaci e gastrointestinali e altre malattie (come ipotiroidismo, celiachia, diabete e leucemia) vengono trattate secondo necessità. Per questo è fondamentale prevedere un approccio multidisciplinare e centrato sulla persona. L’assistenza dei pazienti con sindrome di Down deve includere anche consulenza genetica per la famiglia, assistenza sociale e programmi scolastici appropriati al livello delle funzioni intellettive. Un intervento tempestivo con sistemi educativi e altre forme di assistenza migliora le capacità dei bambini piccoli con sindrome di Down. Lo sviluppo delle persone con sindrome di Down può essere favorito da interventi di supporto mirati sulle esigenze di ognuno. I medici specialisti in pediatria e neuropsichiatra infantile hanno il compito di valutare le specifiche necessità di sviluppo e di definire gli interventi più adeguati. La continuità assistenziale è fondamentale per garantire una buona qualità di vita alle persone con sindrome di Down e alle loro famiglie.
Inclusione Sociale e Autonomia
Il livello di istruzione che la persona con sindrome di Down raggiungerà e l'attività lavorativa che potrà svolgere sono strettamente dipendenti dalle sue personali capacità. Con un supporto, molti adulti sono in grado di condurre una vita attiva con un certo grado di indipendenza. L'accettazione della diagnosi da parte della famiglia può essere rapida o richiedere più tempo. Migliaia di persone in Italia hanno la sindrome di Down e molti genitori trovano utile parlare con altre persone che vivono la loro stessa situazione. È importante trovare un equilibrio tra le attività svolte all’interno della famiglia e quelle specialistiche in maniera tale da garantire che la persona con trisomia 21 cresca in un ambiente adattato alle proprie esigenze, ma è anche utile tener presente che potrà avere bisogno di adattarsi a quello che accade intorno a lui e alle esigenze degli altri membri della famiglia.
Spesso, le persone con sindrome di Down non trovano le condizioni adatte per mettere a frutto il proprio potenziale, e anzi spesso incontrano barriere che ne ostacolano la partecipazione alla vita sociale. Tra le ragioni vi è spesso una scarsa consapevolezza, da parte di molte persone, di che cosa si intenda per inclusione, e molti sanno poco o nulla di questa specifica forma di disabilità e di cosa comporti per le persone colpite. Nel percorso di crescita l’aspetto sociale e pedagogico riveste un ruolo essenziale. Convivere con la sindrome di Down oggi significa affrontare delle sfide ma anche scoprire risorse preziose. I genitori dei ragazzi con Trisomia 21 cercano di favorire l’inclusione sociale e l’autonomia dei propri figli anche attraverso opportunità di inserimento lavorativo: un ambito in cui la nostra società deve ancora compiere passi avanti, superando pregiudizi e mancanza di accoglienza.
