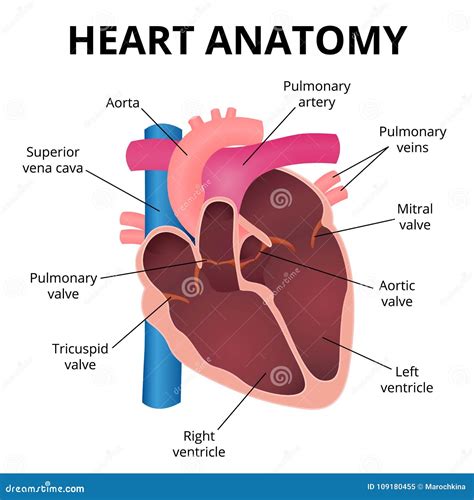
La sindrome del cuore sinistro ipoplasico, nota anche con l’acronimo inglese HLHS (Hypoplastic Left Heart Syndrome), rappresenta una delle malformazioni cardiache congenite più complesse e critiche che possano manifestarsi in ambito neonatale. Questa patologia si configura come un gruppo di difetti strutturali in cui le sezioni sinistre del cuore, essenziali per la circolazione sistemica, risultano gravemente sottosviluppate. In un cuore anatomicamente normale, il lato sinistro - composto dall'atrio sinistro, dal ventricolo sinistro, dalla valvola mitrale, dall'arco aortico e dalla valvola aortica - ha il compito fondamentale di pompare sangue ricco di ossigeno in aorta e, da qui, in tutto l’organismo. Nella HLHS, queste strutture sono troppo piccole o malformate per sostenere tale funzione, rendendo il neonato dipendente da una circolazione alternativa che, se non gestita tempestivamente, conduce inevitabilmente a uno stato di shock e, infine, al decesso.
Eziologia e Sviluppo Embrionale
La sindrome del cuore sinistro ipoplasico si verifica durante la crescita del feto, nel periodo critico in cui il cuore si sviluppa. Dal punto di vista eziologico, la condizione è considerata multifattoriale, derivante da una complessa interazione tra fattori genetici e ambientali. Sebbene la causa precisa rimanga in gran parte sconosciuta, l’ipotesi emodinamica suggerisce che il sottosviluppo del ventricolo sinistro possa essere scatenato da un ridotto flusso di sangue attraverso le sezioni sinistre del cuore durante la vita intrauterina.
Nonostante l'HLHS non segua un modello di ereditarietà semplice, sono state identificate correlazioni con disturbi cromosomici, come la sindrome di Turner, la sindrome di DiGeorge e la sindrome di Noonan. Esistono inoltre evidenze di una predisposizione familiare: il fatto che un fratello nasca con la sindrome del cuore sinistro ipoplastica aumenta il rischio statistico di avere un altro bambino affetto dalla medesima anomalia. Anche l'esposizione materna a determinate sostanze tossiche, l'uso di specifici farmaci durante la gravidanza o condizioni metaboliche materne non controllate sono stati studiati come potenziali fattori di rischio che possono contribuire all'insorgenza di questa anomalia cardiaca.
Fisiopatologia: Il Ruolo Cruciale del Dotto di Botallo
Per comprendere la gravità clinica della HLHS, è necessario analizzare la circolazione fetale. Durante la vita in utero, il feto non utilizza i polmoni per l'ossigenazione; il sangue viene ossigenato dalla placenta e fluisce attraverso connessioni specifiche, tra cui il dotto di Botallo (dotto arterioso), che mette in comunicazione l'arteria polmonare con l'aorta.
Nei neonati affetti da HLHS, il lato sinistro del cuore è incapace di pompare adeguatamente sangue ossigenato in tutto il corpo. Di conseguenza, il lato destro del cuore deve lavorare di più per spingere il sangue sia verso i polmoni che verso il resto dell'organismo. Questa condizione è possibile solo finché il dotto di Botallo rimane aperto. Il dotto di Botallo è destinato a chiudersi fisiologicamente nei primi giorni di vita; quando questo avviene, il neonato va incontro inevitabilmente alla morte, poiché il flusso di sangue verso il corpo diminuisce drasticamente, gli organi vitali vengono ipoperfusi e si instaura uno stato di shock. È vitale, quindi, prevenire la chiusura di questa via di fuga mediante un intervento farmacologico tempestivo.
Diagnosi Fetale e Riconoscimento Clinico
La diagnosi precoce è della massima importanza per la pianificazione terapeutica. Grazie ai progressi dell'ecografia ostetrica, l'HLHS può essere diagnosticato già durante la gravidanza, di solito tra la 20ª e la 24ª settimana di gestazione, attraverso ecografie fetali avanzate. L’ecocardiogramma fetale è l’esame di riferimento che permette di visualizzare dettagliatamente le camere cardiache, confermando le dimensioni ridotte del ventricolo sinistro e dell'aorta.
Qualora la diagnosi non sia stata effettuata in utero, il sospetto clinico sorge poco dopo la nascita, solitamente entro le prime 24-48 ore, quando il dotto arterioso inizia a chiudersi. I sintomi includono respirazione rapida e affannosa, polso debole, pallore o colorazione bluastra della pelle (cianosi), letargia, temperatura corporea bassa e una ridotta produzione di urina. In tali circostanze, la diagnosi deve essere confermata con urgenza mediante un ecocardiogramma neonatale. Il mancato riconoscimento di questi segni o l'esecuzione non corretta di esami diagnostici di follow-up rappresentano criticità che possono influenzare drasticamente la sopravvivenza del piccolo paziente.
Cuore sinistro ipoplasico
Il Percorso Chirurgico: La Ricostruzione Stadiata
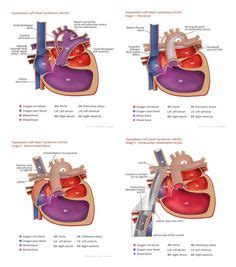
La sopravvivenza del neonato affetto da HLHS richiede un approccio multidisciplinare che prevede una serie di interventi cardiochirurgici finalizzati a separare la circolazione del cuore destro da quella del cuore sinistro. Lo scopo ultimo di queste procedure è consentire al lato destro del cuore di svolgere il lavoro a cui solitamente è preposto il ventricolo sinistro, ovvero pompare il sangue ossigenato in tutto il corpo. Il trattamento chirurgico si articola in tre fasi principali:
Intervento di Norwood (Fase I)
Questa procedura viene solitamente eseguita entro i primi giorni di vita, spesso entro le prime due settimane. È l'operazione più complessa: il chirurgo ricostruisce l'aorta utilizzando l'arteria polmonare e crea una nuova connessione, nota come shunt, per garantire il flusso di sangue ai polmoni. Questo intervento crea una via di deflusso per il sangue ossigenato, unendo la parte inferiore dell’aorta con il ventricolo destro.
Intervento di Glenn (Fase II)
Si esegue, in genere, tra i 4 e i 6 mesi di vita. La finalità di questa fase è chiudere l'accesso della vena cava superiore al cuore e collegarla direttamente all'arteria polmonare. In questo modo, il sangue che ritorna dalla parte superiore del corpo viene indirizzato ai polmoni, evitando di passare attraverso il ventricolo destro, riducendo così il carico di lavoro del cuore.
Intervento di Fontan (Fase III)
Si effettua tipicamente tra i 18 mesi e i 3 anni di età. L'obiettivo è chiudere l'accesso della vena cava inferiore al cuore e collegarla direttamente all'arteria polmonare. A seguito di questo intervento, tutto il sangue venoso povero di ossigeno viene indirizzato verso l'arteria polmonare, permettendo al ventricolo destro di concentrarsi esclusivamente sul pompaggio del sangue ossigenato verso l'organismo.
Gestione Post-operatoria e Prospettive a Lungo Termine
Le prospettive a lungo termine per i bambini con sindrome del cuore sinistro ipoplasico continuano a migliorare, sebbene la condizione rimanga una sfida costante. Dopo gli interventi chirurgici, i pazienti necessitano di un monitoraggio rigoroso che include controlli regolari con ecocardiogrammi e test cardiaci per verificare la funzione cardiaca e lo sviluppo del cuore.
Poiché i pazienti possono essere a rischio di formazione di coaguli sanguigni, soprattutto dopo la fase Fontan, è spesso necessaria una terapia anticoagulante o antipiastrinica a lungo termine, utilizzando farmaci come l'aspirina, il warfarin o l'enoxaparina. In alcuni casi, i bambini devono assumere antibiotici prima delle visite dal dentista e di alcuni interventi chirurgici (come quelli a carico delle vie respiratorie) per prevenire gravi infezioni cardiache chiamate endocarditi.
Il trapianto di cuore, in alcune strutture mediche, è ritenuto la procedura di elezione per la HLHS. Tuttavia, la scarsa disponibilità di donatori cardiaci e i miglioramenti significativi negli esiti chirurgici hanno reso l'approccio multistadio la strategia più comunemente raccomandata. Dopo un eventuale trapianto, i pazienti devono assumere immunosoppressori per il resto della vita, farmaci che rendono più sensibili alle infezioni e aumentano il rischio di sviluppare complicazioni a lungo termine.
Sebbene la sindrome del cuore sinistro ipoplasico rappresenti circa il 25% di tutte le cardiopatie congenite strutturali che richiedono un intervento chirurgico neonatale, l'incidenza stimata varia tra 1 su 3.000 e 1 su 5.000 nati vivi. La ricerca continua a studiare le basi genetiche e ambientali di questa rara patologia, con l'obiettivo di migliorare ulteriormente la qualità e la durata della vita dei bambini nati con questa complessa malformazione cardiaca. La diagnosi precoce, l'assistenza in unità di terapia intensiva neonatale o cardiaca pediatrica e un follow-up costante rappresentano i pilastri fondamentali per gestire questa condizione critica e consentire ai piccoli pazienti di crescere con una funzione cardiaca ottimale.
tags: #ipoplasia #ventricolo #sinistro #fetale