L'igroma cistico, noto anche come linfangioma cistico, rappresenta una rara malformazione congenita del sistema linfatico che insorge durante lo sviluppo embrionale. Questa condizione, caratterizzata dalla formazione di una o più cavità piene di liquido linfatico, pone sfide diagnostiche e terapeutiche significative, ma recenti sviluppi nella medicina fetale e nella ricerca sulle cellule staminali aprono nuove prospettive per una gestione più efficace e per migliorare la prognosi dei nati affetti.
Origine e sviluppo dell’igroma cistico
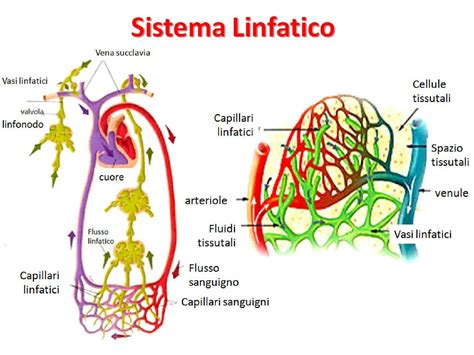
L’origine dell’igroma cistico è legata a un’alterazione del sistema linfatico durante l’embriogenesi. Si ipotizza che derivi da un’insufficiente drenaggio del sistema linfatico in uno dei suoi punti di connessione con il sistema venoso, in particolare a livello dei sacchi linfatici giugulari. Durante lo sviluppo fetale, il sistema linfatico drena nel sacco linfatico giugulare, una struttura primitiva che, intorno ai 40 giorni di età gestazionale, dovrebbe comunicare con la vena giugulare. Se questa comunicazione si interrompe, a causa di un’alterata linfoangiogenesi o di fenomeni ostruttivi, si verifica una dilatazione dei sacchi linfatici laterocervicali, con conseguente formazione dell’igroma cistico. Dal punto di vista istologico, l’igroma cistico è costituito da ampi spazi linfatici rivestiti da cellule endoteliali, separati da uno stroma di tessuto connettivo. Sebbene tecnicamente sia una neoplasia benigna dei vasi linfatici, la sua rilevanza clinica in ambito prenatale è notevole, poiché spesso funge da marcatore per gravi anomalie cromosomiche o sindromi genetiche complesse.
Caratteristiche cliniche e diagnostica prenatale
L’igroma cistico si presenta tipicamente come una tumefazione sottocutanea di consistenza soffice, a decorso generalmente lento. Può essere uni- o multicistico e può interessare diverse parti del corpo, sebbene la localizzazione più frequente sia la regione del collo (circa il 75% dei casi), seguita dalle ascelle. Altre localizzazioni includono il tronco e gli arti. La diagnosi precoce è fondamentale e avviene solitamente nel corso della gravidanza tramite ecografia. L’esame della Translucenza Nucale, eseguito tra la 9ª e la 14ª settimana di gestazione, può rivelare la presenza di questa formazione cistica. In presenza di un igroma cistico, vengono prescritti ulteriori accertamenti per valutare la sua estensione, la presenza di setti interni (forme settate sono statisticamente più correlate ad anomalie del cariotipo) e l’eventuale associazione con altre malformazioni. Le ecografie seriali permettono di monitorare l’evoluzione dell’igroma e l’insorgenza di complicanze come l’idrope fetale, una condizione caratterizzata da un accumulo eccessivo di liquidi in almeno due compartimenti fetali, considerata la più grave complicanza.
Il legame con la Sindrome di Noonan
Una parte dei casi di igroma cistico a cariotipo normale è attribuibile alla sindrome di Noonan. La sindrome di Noonan (SN) è una malattia multisistemica rara molto variabile, caratterizzata prevalentemente da bassa statura, dismorfismi facciali caratteristici, cardiopatie congenite, cardiomiopatia e rischio aumentato di sviluppare tumori durante l’infanzia. È una sindrome caratterizzata da bassa statura per femori corti (inferiori al 3° centile), cardiopatia congenita, persistenza della vena ombelicale di destra, edema nucale, igroma cistico, ascite, idrotorace, idrope non immune, anomalie renali, ipertelorismo, orecchie ad impianto basso e ruotate posteriormente. Di solito la malattia esordisce nel periodo neonatale con difficoltà di alimentazione e ritardo della crescita. Spesso i dismorfismi facciali caratteristici sono maggiormente evidenti nell’infanzia: fronte alta e ampia, ipertelorismo, ptosi palpebrale e rime palpebrali oblique verso il basso, orecchie spesse a impianto basso e ruotate posteriormente, filtro profondo, micrognazia, capelli ricci e collo corto, talvolta con pterigio.
Eziologia e basi genetiche delle RASopatie
Le RASopatie costituiscono uno tra i più comuni gruppi di patologie dello sviluppo a base non cromosomica con una prevalenza complessiva di 1/1500 nuovi nati. Fra le RASopatie, la sindrome di Noonan rappresenta la condizione più variabile dal punto di vista clinico e a più alta incidenza. Queste malattie dello sviluppo comprendono anche la sindrome cardio-facio-cutanea (CFCS), la sindrome di Costello (CS), la sindrome di Noonan con lentiggini multiple (NSML), la sindrome di Mazzanti (MS), la neurofibromatosi tipo 1 (NF1), la sindrome di Legius (LS) e un numero crescente di nuove condizioni clinicamente e molecolarmente correlate. Le RASopatie condividono un meccanismo patogenetico comune caratterizzato da anomalie della regolazione della via di trasduzione del segnale mediata dalle proteine RAS. La sindrome di Noonan è causata da mutazioni dei geni PTPN11 (12q24.13), presenti nel 50% dei pazienti, SOS1 (2p22.1) nel 15%, RAF1 (3p25.2), RIT1 (1q22) e LZTR1 (22q11.21) e, più raramente, di altri geni associati alla via di segnale di RAS/MAPK.
Recettori tirosina chinasi e segnalazione MAP chinasi (via MAPK)
Il gene PTPN11 è il responsabile della produzione di una proteina chiamata SHP-2, fondamentale per la trasmissione di diversi segnali intracellulari e per il controllo dello sviluppo del mesodermico e della genesi della valvola cardiaca semilunare. Le mutazioni di PTPN11 presenti nella SN danno luogo a un aumento della proteina SHP-2 e a un’alterata regolazione delle vie di trasduzione dei segnali cellulari attivati dalla proteina Ras e dalle protein-chinasi attivate da mitogeni (MAPK). La via di segnalazione Ras-MAPK produce segnali per i fattori di accrescimento e citochine che partecipano nella trascrizione dei geni coinvolti nella proliferazione cellulare e nella determinazione della differenziazione cellulare.
Spettro clinico e manifestazioni multisistemiche
Il fenotipo della Sindrome di Noonan è caratterizzato da bassa statura, ipertelorismo, impianto basso delle orecchie che appaiono anche ruotate posteriormente, collo tozzo con cute ridondante o con pieghe laterali, pieghe epicantiche, sordità, ritardo motorio, difetti della coagulazione per trombocitopenia. La cardiopatia congenita è nel 40-50% dei casi rappresentata da stenosi polmonare, o da cardiomiopatia ipertrofica (20-30%), DIV, DIA, Tetralogia di Fallot. I pazienti hanno difficoltà a guadagnare peso e molti di essi mantengono una corporatura snella per tutta la vita. I principali segni clinici ortopedici comprendono la deformità dello sterno, i piedi equino-vari e la scoliosi progressiva (che esordisce nell’adolescenza). Spesso la pelle è secca e, talvolta, ipercheratosica sulle mani e sui piedi. I pazienti possono presentare linfedema periferico, che in alcuni può essere progressivo ed esteso. Sono comuni le anomalie oculari (strabismo, errori refrattivi) e l’affastellamento dei denti. Il 10% dei pazienti presenta sordità.
Diagnosi differenziale e complessità clinica
La diagnosi si basa sull’osservazione del quadro clinico, ma può essere difficile a causa della sua notevole variabilità. Nell’età adulta il dimorfismo facciale si riduce; restano però un solco nasolabiale prominente e uno pterigio del collo. Inizialmente la sindrome descritta da Jacqueline Noonan fu definita erroneamente “sindrome di Turner maschile”. Tuttavia, la sindrome di Noonan è un’entità clinica distinta. La diagnosi differenziale di questa sindrome include: la sindrome di Turner, la sindrome di Leopard, la sindrome cardio-facio-cutanea e la sindrome di Costello. Nel 50% dei pazienti è presente una cardiopatia congenita. I difetti più frequenti sono la stenosi valvolare polmonare e la cardiomiopatia ipertrofica. Più raramente possono essere diagnosticati altre cardiopatie. Nei primi mesi di vita sono frequenti i problemi alimentari, che comprendono la semplice difficoltà alla suzione fino alle anomalie più complesse che rendono necessario il ricorso a tecniche di alimentazione artificiale.
Prospettive nella medicina fetale
La medicina fetale, che riconosce al feto la dignità di paziente a tutti gli effetti, ha rivoluzionato la percezione e la gestione di patologie come l’igroma cistico. Studi condotti hanno dimostrato che, in una percentuale significativa di casi, l’igroma cistico non è necessariamente associato a malattie genetiche o cromosomiche con prognosi infausta. In uno studio su 220 gravidanze con diagnosi di igroma cistico, è emerso che nel 55-59% dei casi i cromosomi fetali erano normali. Ulteriori indagini, come l’ecocardiografia fetale, hanno escluso patologie cardiache nel 70% dei casi. Questi studi hanno evidenziato che la regressione intrauterina dell’igroma cistico si osserva nel 36% dei casi, con una completa scomparsa nel 77%.
Gestione terapeutica e multidisciplinare
Il trattamento dell’igroma cistico mira a ridurre le dimensioni delle cisti e ad alleviare i sintomi associati. L’asportazione chirurgica della massa rimane la modalità di trattamento primaria, anche se la recidiva è un problema comune. La scleroterapia, consistente nell’iniezione di sostanze irritanti direttamente nelle cisti per indurne il collasso e la successiva fibrosi, è un approccio meno invasivo. Per quanto riguarda la sindrome di Noonan, il trattamento richiede un approccio multidisciplinare. Le anomalie cardiovascolari vengono trattate secondo i protocolli standard. Il trattamento del ritardo della crescita con l’ormone della crescita rimane controverso. La prognosi è variabile dato che il quadro clinico comprende i segni clinici lievi o non evidenti nell’età adulta fino alle forme più gravi associate a cardiopatie potenzialmente letali o a tumori maligni infantili. Utilizzando trattamenti appropriati, la maggior parte delle persone con sindrome di Noonan conduce una vita adulta normale.
Ricerca futura e frontiere terapeutiche
La collaborazione tra istituti di ricerca sta aprendo nuove frontiere nel trattamento delle patologie fetali, inclusi gli igromi cistici. La ricerca sulle cellule staminali mesenchimali amniotiche e sulle staminali ematopoietiche del cordone ombelicale sta esplorando il loro potenziale uso come terapia fetale per malattie genetiche. L’obiettivo è trapiantare il feto in una fase molto precoce dello sviluppo, quando la sua reazione immunitaria è ancora bassa o inesistente, per consentire la nascita di bambini sani. Studi su modelli animali hanno dimostrato la capacità di queste cellule di migrare e attecchire negli organi ematopoietici e in altri tessuti, aprendo la strada a future applicazioni terapeutiche. Inoltre, si stanno sperimentando approcci terapeutici prenatali specifici per l’igroma cistico. Esperienze condotte inoculando sostanze come l’OK-432 direttamente all’interno dell’igroma cistico hanno mostrato una percentuale di sopravvivenza promettente. Più recentemente, nella fase postnatale, farmaci come il Sirolimus hanno dimostrato una significativa riduzione del volume di voluminosi igromi cistici.