La sindrome di Down, nota anche come Trisomia 21, è una condizione genetica complessa che incide profondamente sullo sviluppo fisico e cognitivo degli individui. Sebbene sia ampiamente riconosciuta, la comprensione delle sue cause, delle metodologie diagnostiche e dei percorsi di ricerca in atto rimane fondamentale per promuovere una maggiore inclusione e offrire il miglior supporto possibile alle persone che ne sono affette e alle loro famiglie. Questa condizione, che accompagna la specie umana senza differenze significative tra i sessi e le etnie, è stata oggetto di studio e sensibilizzazione crescenti, con l'obiettivo di abbattere barriere e pregiudizi.
Comprendere la Sindrome di Down: Dalla Scoperta alle Caratteristiche Fondamentali
La sindrome di Down è una condizione causata dalla presenza di tre copie del cromosoma 21, anziché due, in tutte le cellule dell’organismo. Questo cromosoma extra, presente in tutte le cellule del corpo, altera il normale sviluppo fisico e cognitivo, determinando una disabilità intellettiva di varia gravità e altri problemi di salute. Le cellule umane contengono in genere 46 cromosomi organizzati in 23 coppie numerate. La sindrome è detta anche trisomia 21, proprio a indicare lo specifico cromosoma coinvolto, nonché la terza copia del cromosoma, all’origine dei problemi di sviluppo e delle tipiche caratteristiche fisiche delle persone Down. Non tutti i difetti sono presenti in ciascuna persona malata, e le persone con sindrome di Down presentano gradi variabili, da lieve a grave, di disabilità intellettiva e fisica.
1.1. Breve Storia: Il Contributo di John Langdon Down
La sindrome di Down prende il proprio nome dal medico britannico che per primo la studiò a fondo, fino a identificarne i tratti comuni nelle persone affette. Si chiamava John Langdon Down e fu l’autore, nel 1866, di una pubblicazione scientifica in cui si descriveva in maniera completa la sindrome. Down aveva incontrato per la prima volta una ragazza con disabilità dello sviluppo quando lei aveva circa 18 anni. Lui stava facendo una passeggiata con la sua famiglia in campagna e aveva cercato riparo dalla pioggia in una fattoria nei paraggi. L’incontro con la ragazza lo colpì così tanto da decidere, in quel momento, di voler studiare medicina e fare della cura dei disabili il lavoro della propria vita.
1.2. Prevalenza e Aspettativa di Vita: Uno Sguardo ai Numeri
L’incidenza di nati vivi con la sindrome a livello mondiale è di circa uno su 1.000, mentre l’incidenza varia, a seconda della zona geografica, da 1 su 319 bambini a 1 su 1.000. Negli Stati Uniti d’America la media è di un nato su 700. In Italia, secondo alcune stime, circa un bambino ogni 1.200 nati ha la sindrome: si tratterebbe di circa 500 nascite all’anno per un totale di 38.000 persone nel nostro Paese. A livello globale, la Sindrome di Down si verifica in un caso su 1.000-1.200 nati vivi.
1.3. L'Importanza dell'Inclusione Sociale
Esistono da tempo numerose occasioni di sensibilizzazione sulla sindrome di Down, in cui si cerca di affrontarne diversi aspetti, cercando di chiarire i dubbi e aumentare le conoscenze. L’obiettivo è favorire, per le persone con la sindrome di Down, una maggior inclusione nella società. Le persone con sindrome di Down hanno infatti tutte le “carte in regola” per realizzarsi, sul lavoro così come nella vita privata. Spesso però non trovano le condizioni adatte per mettere a frutto il proprio potenziale, e anzi spesso incontrano barriere che ne ostacolano la partecipazione alla vita sociale. Tra le ragioni vi è spesso una scarsa consapevolezza, da parte di molte persone, di che cosa si intenda per inclusione. Inoltre molti sanno poco o nulla di questa specifica forma di disabilità e di cosa comporti per le persone colpite. Nel percorso di crescita l’aspetto sociale e pedagogico riveste un ruolo essenziale. Convivere con la sindrome di Down oggi significa affrontare delle sfide ma anche scoprire risorse preziose. I genitori dei ragazzi con Trisomia 21 cercano di favorire l’inclusione sociale e l’autonomia dei propri figli anche attraverso opportunità di inserimento lavorativo: un ambito in cui la nostra società deve ancora compiere passi avanti, superando pregiudizi e mancanza di accoglienza.
Persone affette da sindrome di down, "Chi trova un lavoro trova un tesoro"
Le Radici Genetiche della Sindrome di Down: Cause e Tipologie
Le strutture in cui il materiale genetico si aggrega all’interno delle nostre cellule sono i cromosomi. Il nucleo di ciascuna cellula umana contiene normalmente 46 cromosomi, suddivisi in 23 coppie. Ogni coppia è formata da un cromosoma ereditato dalla madre e da uno ereditato dal padre. Nelle persone con sindrome di Down, il processo di divisione cellulare al momento del concepimento è avvenuto in modo anomalo e per questo i cromosomi numero 21 sono presenti in tre copie anziché due. Perché si verifichi quest’anomalia non è chiaro. Si tratta di una condizione che accompagna la specie umana. La sindrome di Down si presenta quando, prima o dopo il concepimento, si verificano delle anomalie durante il meccanismo di separazione dei cromosomi. Il motivo non è legato a una causa particolare poiché è un evento spontaneo, tranne in rari casi.
2.1. Il Ruolo dei Cromosomi: La Trisomia 21 "Libera"
In circa il 95% dei casi, la sindrome di Down è causata da una non disgiunzione con conseguente formazione di un cromosoma 21 supplementare (trisomia 21), che è tipicamente di origine materna. La non disgiunzione si riferisce al mancato corretto distacco dei cromosomi omologhi o dei cromatidi fratelli durante la meiosi dei gameti. Le persone affette hanno 47 cromosomi invece dei normali 46. Questo è il tipo più comune e si definisce "libera" ed è influenzata dall'età materna. Questa forma è causata da un particolare errore nella divisione cellulare: prima o durante il concepimento, una coppia di cromosomi 21, che si trovi nello spermatozoo o nell’uovo, non riesce a separarsi. Di conseguenza l’embrione inizia a formarsi con tre copie del cromosoma 21 anziché le consuete due. La trisomia 21 libera, ha origine da un errore casuale che può avvenire sia durante lo sviluppo della cellula uovo della madre che dello spermatozoo, qualunque sia l’età dei genitori; questo errore casuale avviene però più frequentemente nella cellula uovo con l’aumentare dell’età della donna. Per i genitori e i familiari di un bambino con trisomia 21 libera, il rischio di ricorrenza non aumenta in modo significativo rispetto a quello della popolazione generale.
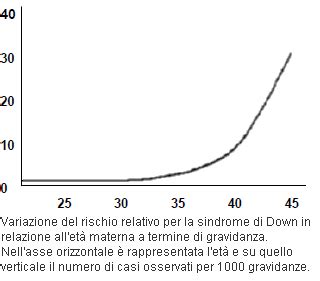
2.2. L'Età Materna come Fattore di Rischio: Comprendere la Correlazione
Un fattore di rischio nell’avere un figlio con sindrome di Down è l’età materna al momento del concepimento: in particolare il rischio sembra aumentare dai 35 anni in su. L’incidenza aumenta con l'avanzamento dell'età materna, anche se non si sono mai dimostrate le cause di questa relazione. Stando ai dati pubblicati dall’Istituto superiore di Sanità, numerose indagini epidemiologiche hanno messo in evidenza che l'incidenza aumenta con l'avanzamento dell'età materna.
2.3. Altre Forme della Sindrome: Traslocazione e Mosaicismo
Oltre alla trisomia 21 libera, esistono altre tipologie meno comuni. Circa il 4% dei casi di sindrome di Down è dovuto a una traslocazione genetica. In una traslocazione bilanciata, il materiale genetico viene scambiato con materiale proveniente da un altro cromosoma non-omologi, e la conta cromosomica viene mantenuta a 46. In una traslocazione sbilanciata, c'è un guadagno o una perdita di materiale genetico, che crea uno squilibrio genetico e successive anomalie cliniche. La traslocazione più comune è t(14;21), in cui il cromosoma 21 è attaccato al cromosoma 14; questa è una traslocazione sbilanciata, con conseguente conta cromosomica di 45. In circa la metà dei soggetti con t(14;21) entrambi i genitori hanno un cariotipo normale, mostrando una traslocazione insorta de novo. Nell'altra metà, un genitore (quasi sempre la madre), che non ha la sindrome di Down, ha solo 45 cromosomi, uno dei quali è t(14;21). In teoria, la probabilità che una madre portatrice possa avere un bambino con sindrome di Down è di 1:3, ma il rischio reale è inferiore (circa 1:10). Se il padre è il portatore, il rischio è solo di 1:20. Un'altra tra le traslocazioni più frequenti è t(21;22). In questi casi, le madri portatrici hanno un rischio di circa 1:10 di avere un bambino con la sindrome di Down; il rischio è ancora minore per i padri portatori.
La traslocazione 21q21q, che si verifica quando il cromosoma 21 è collegato a un altro cromosoma 21, è molto meno comune. Determinare se un genitore è portatore o mosaico per la traslocazione 21q;21q è importante per comprendere i rischi per i loro figli. Un genitore portatore (ovvero, uno che ha una traslocazione bilanciata) ha una probabilità del 100% di avere un figlio affetto dalla sindrome di Down perché tutta la prole vitale avrebbe la sindrome di Down o la monosomia 21. La monosomia 21 è tipicamente incompatibile con la vita.Nel caso della sindrome di Down da traslocazione il rischio di ricorrenza aumenta considerevolmente se uno dei genitori è portatore della traslocazione in forma bilanciata, (ovvero senza perdita o aggiunta di materiale genetico nel suo DNA) indipendentemente dall’età. Anche i fratelli e le sorelle di un bambino con sindrome di Down da traslocazione possono essere portatori della traslocazione bilanciata, e avere quindi un rischio aumentato per le loro gravidanze. Solo nel caso in cui uno dei due genitori sia portatore di traslocazione è possibile che la Trisomia 21 venga trasmessa al bambino.
Il mosaicismo della sindrome di Down presumibilmente risulta da non-disgiunzione durante la divisione cellulare nell'embrione. Di più raro riscontro (2% dei casi), è caratterizzato dalla contemporanea presenza, in percentuali diverse da individuo a individuo, di cellule con corredo cromosomico a 46 cromosomi e a 47 cromosomi. Le persone con mosaicismo della sindrome di Down hanno 2 linee cellulari: una con i normali 46 cromosomi e una con 47 cromosomi, tra cui un cromosoma 21 in più, o un diverso numero di cromosomi, a seconda della traslocazione. Sebbene il loro rischio di avere un bambino con sindrome di Down sia notevolmente aumentato, il genitore può anche avere figli con cromosomi normali. Nelle persone con mosaicismo della sindrome di Down, la prognosi per l'intelligenza e il rischio di complicanze mediche probabilmente dipende dalla proporzione di cellule anormali (p. es., trisomia 21) in ciascun tessuto diverso, compreso il cervello. Tuttavia, in pratica, questo rischio non può essere previsto in quanto non è possibile determinare il cariotipo in ogni singola cellula del corpo. Alcune persone con mosaicismo della sindrome di Down hanno segni clinici molto sottili e possono avere un'intelligenza normale; tuttavia, anche le persone con sindrome di Down senza mosaicismo possono avere reperti clinici variabili. Questo vale anche per le persone affette che non sono mosaiche.
2.4. Fattori di Rischio Ulteriori: L'Influenza Paterna e Ambientale
Esiste un rischio legato all’età materna, ma alla luce degli studi e delle indagini sempre più approfondite, non è esclusa nessuna fascia di età ed esiste anche un fattore legato all’età paterna, del quale si parla poco, oltre a una multifattorialità. Nonostante non sia stata molto approfondita, esiste anche un’influenza, seppur inferiore (5-6%), dell’età paterna sulla possibilità di avere un bambino con sindrome di Down.Altri fattori che sono stati correlati a un aumentato rischio, sebbene con gradi di evidenza variabili, includono:
- Fumo e consumo di prodotti a base di nicotina: Questi possono influire sulla salute generale e sulla stabilità genetica.
- Consumo di alcolici: Riduce nel tempo l’assorbimento di vitamina B12 e di acido folico, di più nel caso di una mutazione del gene MTHFR, che regola il metabolismo dei folati.
- Status socio-economico: Uno studio condotto in diversi stati americani ha osservato che la bassa scolarizzazione familiare e uno status economico precario possono incidere sul maggiore tasso di Trisomia 21. Probabilmente perché ciò comporta un accesso più complicato ai programmi sanitari di screening e una minore consapevolezza verso uno stile di vita più salutare.La presenza di sindrome di Down in familiari di primo grado aumenta leggermente il rischio di avere un bambino con la stessa condizione.
Diagnosi della Sindrome di Down: Dallo Screening Prenatale alla Conferma Post-natale
Durante la gravidanza non esistono sintomi specifici nella madre che possano indicare la presenza della sindrome di Down nel feto. Per questo motivo, la diagnosi si avvale di una serie di test di screening e diagnostici, che possono essere eseguiti sia prima che dopo la nascita. Lo screening prenatale per la sindrome di Down è possibile con criteri di assoluta certezza solo con lo studio del cariotipo fetale effettuabile mediante amniocentesi ovvero mediante prelievo di villi coriali, mentre gli altri possibili accertamenti (privi di concreto pericolo per la gestante e per il feto) forniscono unicamente una più o meno accurata stima del rischio. Tutti i centri di screening dovrebbero essere in grado di disporre di un adeguato servizio di counselling pre e post test. L'ecografia fetale e i test sierici materni, così come lo screening prenatale non invasivo, vengono offerti a tutte le donne incinte in contesti con elevate risorse come test di screening per anomalie cromosomiche, inclusa la sindrome di Down.
3.1. Screening Non Invasivi: Valutare le Probabilità
Screening non invasivi possono essere condotti già nei primi mesi di gravidanza per calcolare le probabilità che il feto abbia la sindrome di Down.
Test Combinato del Primo Trimestre (Bi-Test e Translucenza Nucale)
Lo screening combinato per la Sindrome di Down al 1° trimestre (11-14 settimane gestazionali) si è sempre più affermato. È più precoce rispetto al tritest eseguito al II trimestre, e più sensibile grazie alla combinazione con la translucenza nucale (NT), marcatore ecografico importantissimo. Lo screening da una stima del rischio (in termini di probabilità statistica) di avere un bambino affetto da Cromosomopatie. Per elaborare tale rischio serve la misurazione della NT del feto tramite ecografia eseguita da operatori esperti, la quale, assieme alla determinazione di due parametri biochimici come la Free beta HCG e la PAPP-A, permette di arrivare ad una sensibilità di circa il 90%, ben più alta di quella consentita dallo screening TRITEST al 2° trimestre (attorno al 70%).
Il BI-TEST può essere eseguito sul sangue materno alla fine del primo trimestre di gravidanza (12ª settimana di gestazione) dosando due proteine: la free-beta hCG (frazione libera della gonadotropina corionica) e la PAPP-A (proteina A plasmatica associata alla gravidanza). La PAPP-A è una glicoproteina, rilasciata dalla placenta, con alto Peso Molecolare (720-850 KDa) che appartiene alla famiglia delle metalloproteinasi zinco dipendenti ed è stata identificata per la prima volta nel plasma delle donne in gravidanza all'inizio degli anni '70. Durante la gravidanza, la concentrazione di PAPP-A aumenta costantemente dalla 7a settimana fino al parto. Dopo il parto il livello diminuisce rapidamente con una emivita di 3-4 giorni. Il livello di hCG aumenta rapidamente nelle prime due settimane dopo il concepimento, raggiungendo il picco massimo durante la 9ª settimana, per poi diminuire gradualmente durante il 2° e il 3° trimestre. Dall’integrazione tra questi valori e l’età della madre è possibile individuare il 65% dei feti affetti da sindrome di Down, con un 5% di falsi positivi. A questo test si associa un’ecografia fra la 11ª e la 13ª settimana di gravidanza, che valuta lo spessore di uno spazio liquido che si trova in corrispondenza della nuca fetale (translucenza nucale), che pure consente di individuare circa il 75% dei casi di sindrome di Down. L'ecografia fetale può rilevare anomalie come un aumento della translucenza nucale.
Test del DNA Fetale (NIPT)
Lo screening prenatale non invasivo (NIPT Test), in cui il DNA fetale privo di cellule proveniente dalla circolazione materna viene testato per l'aneuploidia, è ora l'opzione di screening di scelta per la trisomia 21 in ambienti ad alta risorsa perché ha una buona sensibilità e specificità. Ricerca il DNA fetale nel sangue materno e può essere eseguito a partire dalla decima settimana di gestazione. Si tratta di un esame molto sofisticato con una sensibilità e una specificità che si avvicinerebbero al 100%. È un test non invasivo molto affidabile, che analizza il DNA fetale libero nel sangue materno attraverso un prelievo ematico. Può essere eseguito a partire dalla 10^ settimana di gestazione in poi, per valutare il rischio di diverse trisomie, tra cui la sindrome di Down, per cui l’accuratezza sale quasi al 99%. In Italia fino ad oggi è stato poco utilizzato in quanto molto costoso e non dispensato dal Servizio Sanitario Nazionale, tranne in alcune regioni dove è stato inserito nei livelli essenziali di assistenza (Emilia Romagna, Valle d’Aosta).
Tri-Test nel Secondo Trimestre
Il TRITEST può essere eseguito tra la 15ª e la 17ª settimana di gestazione. I dosaggi sul siero materno possono mostrare livelli anormali di alfa-fetoproteina, beta-hCG (gonadotropina corionica umana), estriolo non coniugato e inibina nei primi mesi del 2o trimestre (tra la 15a e la 16a settimana di gestazione). Questo dosaggio viene utilizzato nel 2° trimestre di gravidanza per lo screening della Trisomia 21 nel cosiddetto TRITEST. In questo caso il dosaggio viene associato ad altri dati clinici e biologici, come il livello di AFP, l'Estriolo non coniugato, l'età materna e l'epoca gestazionale. Durante la gravidanza, l'estriolo viene prodotto a livello placentare da precursori sintetizzati nella ghiandola surrenale fetale. La forma principale misurata è l'estriolo non coniugato (uE3). In una gravidanza non complicata, i livelli nel siero materno di estriolo totale e non coniugato aumentano nel corso di tutta la gravidanza fino al termine. Nelle gravidanze affette da trisomia 21, il livello di estriolo non coniugato nel siero è ridotto di circa il 25-30%. L'ecografia morfologica, eseguita tra la 16^ e la 18^ settimana, può rilevare delle caratteristiche congenite legate alla sindrome di Down, ma non confermarla senza un test combinato o un test del DNA fetale a precedere.
I risultati dei test di screening sopracitati danno comunque solo risposte di tipo probabilistico.
3.2. Diagnosi Invasiva: Certezza Attraverso l'Analisi del Cariotipo
Se la sindrome di Down è sospettata sulla base di test di screening del siero materno, ecografia o screening prenatale non invasivo, si raccomanda un test di conferma fetale o postnatale. Per l’accertamento sono poi possibili esami ulteriori, invasivi ma che forniscono informazioni più precise, consigliate soprattutto per donne con più di 35 anni. I metodi di conferma fetali comprendono il prelievo dei villi coriali e/o l'amniocentesi con analisi del cariotipo come test di scelta, ma la conferma può essere fatta anche con FISH e con l'analisi cromosomica con microarray.
Villocentesi
La villocentesi, o prelievo dei villi coriali, analizza i cromosomi fetali attraverso il prelievo di cellule della placenta. Viene effettuata intorno alla 15ª/19ª settimana di gestazione.
Amniocentesi
L’amniocentesi esamina una piccolissima quantità di liquido amniotico dopo averlo aspirato con un ago dall’utero materno. Consiste nel prelievo di circa 15-30 ml di liquido amniotico, attraverso un ago che viene introdotto nell’addome materno, sotto monitoraggio ecografico per evitare il più possibile danni al feto.
Cordocentesi
Il prelievo percutaneo di sangue dal cordone ombelicale, noto anche come cordocentesi, è una procedura invasiva utilizzata per ottenere sangue fetale direttamente dal cordone ombelicale, consentendo un cariotipo rapido e la conferma della trisomia 21. La cordonocentesi si effettua dopo la 18ª settimana di gestazione. Il prelievo percutaneo di sangue cordonale è in genere riservato a situazioni in cui altri metodi diagnostici non sono conclusivi o quando sono necessari risultati rapidi.
3.3. Diagnosi alla Nascita e Conferma Post-Natale
Al momento della nascita, per diagnosticare la sindrome di Down si osservano le caratteristiche fisiche del neonato, tra cui la forma del capo o il peso e le dimensioni ridotte rispetto alla media, per esempio. La diagnosi post-natale nei neonati si basa sull’osservazione delle caratteristiche cliniche, di sintomi legati a patologie congenite già diagnosticate in gravidanza, di sintomi nuovi, e rende necessaria una presa in cura multidisciplinare. Un’ulteriore conferma avviene attraverso un test genetico a partire dal sangue del neonato, che permette di definire in modo inequivocabile la presenza della copia extra di cromosoma 21 nelle cellule. Viene eseguito anche il test della conferma del cariotipo, per il numero dei cromosomi, e viene valutata la possibile coesistenza di altre patologie genetiche o congenite, non diagnosticabili nel periodo prenatale.
Manifestazioni Cliniche e Complicazioni Associate alla Sindrome di Down
Come la maggior parte delle condizioni che derivano da uno squilibrio cromosoma, la sindrome di Down colpisce più sistemi e causa sia difetti strutturali che funzionali. Sebbene ogni persona con sindrome di Down sia unica, vi sono alcune caratteristiche fisiche e problematiche di salute comuni.
4.1. Caratteristiche Fisiche Distintive
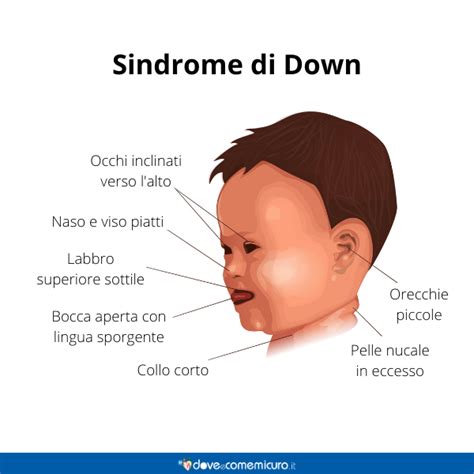
Oltre a caratteristiche uniche e soggettive, alcuni tratti dell’aspetto fisico sono riconosciuti come comuni a tutte le persone con questa sindrome. I neonati affetti tendono a essere tranquilli, piangono raramente e presentano ipotonia. La maggior parte ha un profilo facciale piatto (soprattutto appiattimento del ponte nasale), ma alcuni non hanno evidenti caratteristiche fisiche inusuali alla nascita e successivamente sviluppano durante l'infanzia le caratteristiche facciali più evidenti. Sono frequenti occipite appiattito, microcefalia ed eccesso di cute nella parte posteriore del collo. Il cranio è di norma piccolo e potrebbe presentare un appiattimento a livello occipitale; le fontanelle, nei neonati, sono larghe e si chiudono in ritardo rispetto alla norma.Le caratteristiche fisiche tipiche comprendono le seguenti:
- Gli occhi hanno un'angolazione diretta verso l'alto sul bordo laterale, di solito sono presenti pieghe epicantali agli angoli interni, e potrebbero essere visibili le macchie di Brushfield (macchie grigie o bianche simili a grani di sale, situate perifericamente rispetto all'iride).
- La bocca è spesso mantenuta aperta; una lingua protrusa, rugosa può non avere una scissura centrale. Le orecchie sono spesso piccole e arrotondate.
- Le mani sono spesso corte e larghe e spesso hanno una sola piega palmare trasversale, e le dita sono spesso corte, con clinodattilia (incurvamento) del 5° dito, che spesso ha solo 2 falangi.
- Il piede può presentare un'ampia distanza fra il primo e il secondo dito (dita a sandalo), e il solco plantare spesso si prolunga posteriormente sul piede.La statura è generalmente ridotta e lo sviluppo è più lento rispetto alla media; inoltre il tono muscolare è scarso e le articolazioni sono spesso caratterizzate da lassità. La statura è spesso bassa.

4.2. Sviluppo Cognitivo e Motorio
Chi ha la sindrome di Down presenta gradi variabili, da lieve a grave, di disabilità intellettiva e fisica. La maggior parte delle persone affette ha un grado di deterioramento cognitivo, da grave (QI 20-35) a lieve (QI 50-75). Il quoziente intellettivo ha ampia variabilità intra-sindromica: raramente è normale e generalmente è compreso tra 50 e 80. Ritardi motori e del linguaggio sono evidenti anche in giovane età. Il linguaggio è la funzione strettamente legata allo sviluppo cognitivo che appare più compromessa rispetto all’organizzazione delle altre abilità superiori e al livello intellettivo. I sintomi della patologia cardiaca sono determinati dal tipo ed entità dell'anomalia cardiaca.Quando i bambini affetti crescono, il ritardo fisico e lo sviluppo mentale diventano evidenti. Spesso durante l'infanzia il comportamento suggerisce disturbi di attenzione e iperattività e l'incidenza del comportamento associato al disturbo dello spettro autistico risulta aumentata (in particolare nei bambini con grave disabilità intellettiva). Si possono presentare disturbi del linguaggio, iperattività e difficoltà dell’attenzione, ma ad oggi, grazie anche a una diagnosi precoce e a programmi sempre più su misura, sia educativi che riabilitativi, molti bambini acquisiscono buone competenze linguistiche e mostrano continui progressi nell’apprendimento. Anche le caratteristiche caratteriali e comportamentali sono variabili, dal momento che possono cambiare in relazione all’età e allo stato di salute generale del bambino, poi adulto. Il carattere affettuoso e la socialità possono alternarsi a momenti di rigidità e ostinazione (soprattutto durante la crescita), rabbia e scatti d’ira, depressione e ansia. Vi è un aumentato rischio di depressione nei bambini e negli adulti con sindrome di Down.
4.3. Comorbidità Mediche Frequenti: Un Approccio Multidisciplinare
Alcune patologie sono più frequenti in chi ha questa anomalia cromosomica. Per questi motivi è fondamentale che le persone con sindrome di Down seguano abitudini e comportamenti salutari e si sottopongano a controlli puntuali, di modo che i segni e sintomi precoci di complicanze possano essere colti tempestivamente e gli interventi e l’assistenza medica possano essere immediati e personalizzati. La condizione è tuttavia permanente, può essere curata ma non guarita.
Cardiopatie Congenite
È il caso delle cardiopatie congenite, che riguardano più o meno la metà delle persone con sindrome di Down, e possono essere gravi al punto da richiedere controlli periodici e interventi chirurgici già nella prima infanzia. Cardiopatie congenite interessano 20-76% dei nati vivi con sindrome di Down (6-8% popolazione generale). Nel 50% circa dei neonati affetti è presente una cardiopatia congenita; i difetti del setto interventricolare e i difetti del setto atrioventricolare (chiamati anche difetto dei cuscinetti endocardici o difetti del canale atrioventricolare) sono i più frequenti. I neonati con difetti cardiaci congeniti, i più comuni dei quali sono difetti del setto interventricolare e difetti del setto atrioventricolare, possono essere asintomatici o mostrare segni di insufficienza cardiaca (p. es., respiro affannoso, aumentata frequenza respiratoria, difficoltà di alimentazione, sudorazione, scarso aumento di peso). Soffi possono non essere sempre presenti; tuttavia, sono possibili un certo numero di soffi differenti.
Problemi Gastrointestinali ed Endocrini
Sono frequenti anche i disturbi a carico dell’apparato gastrointestinale, con vari gradi di intensità. Circa il 6% delle persone affette ha anomalie gastrointestinali, in particolare atresia duodenale, talvolta con pancreas anulare. Anche la malattia di Hirschsprung e la celiachia sono più comuni. I neonati con malattia di Hirschsprung di solito hanno un ritardo nell'evacuazione del meconio. I neonati gravemente colpiti possono avere segni di occlusione intestinale (p. es., vomito biliare, mancata progressione di feci, distensione addominale). Atresia o stenosi duodenale possono manifestarsi con vomito biliare o senza sintomi, a seconda dell'entità della stenosi. Questi difetti possono essere rilevati mediante l'ecografia prenatale (segno della doppia bolla).Molte persone sviluppano endocrinopatie, tra cui malattie della tiroide (il più delle volte ipotiroidismo) e diabete.
Compromissioni Sensoriali e Altri Rischi
Sono frequenti anche i disturbi immunitari, come per esempio un aumentato rischio di sviluppare malattie autoimmuni e una maggiore sensibilità alle infezioni. La sindrome di Down aumenta anche la suscettibilità a determinati tipi di cancro, come alcune forme di leucemia durante l’infanzia.Chi soffre della sindrome ha inoltre una maggiore tendenza a sviluppare obesità rispetto al resto della popolazione, può avere problemi spinali, come per esempio un disallineamento delle vertebre. L'ipermobilità atlanto-occipitale e atlanto-assiale, così come anomalie ossee della colonna cervicale, causano instabilità atlanto-occipitale e cervicale; possono verificarsi debolezza e paralisi.Circa il 60% delle persone ha problemi agli occhi, tra cui cataratte congenite, glaucoma, strabismo ed errori di rifrazione; tuttavia, questa prevalenza può variare tra le diverse popolazioni. La maggior parte delle persone ha una perdita dell'udito, e le infezioni dell'orecchio sono molto frequenti.
La Ricerca Scientifica sulla Sindrome di Down: Orizzonti e Progressi
La ricerca sulla sindrome di Down è attiva su molti fronti. Si studiano per esempio i meccanismi molecolari con cui l’anomalia genetica esercita i suoi effetti. Inoltre si cercano metodi per rendere più precisa la diagnosi, si approfondiscono i meccanismi patologici e si cercano nuovi trattamenti per le comorbidità (che ricordiamo sono a carico di molti organi e sistemi). C’è poi la possibilità (che per ora è soprattutto un sogno) di sviluppare una vera e propria cura. Questi obiettivi sono perseguiti studiando l’anomalia anche e i suoi possibili rimedi in animali di laboratorio, studiando gli effetti sia di geni specifici sia di gruppi di geni che possono essere coinvolti nella sindrome.
5.1. Studio dei Meccanismi Molecolari e Genetici
L’ipotesi iniziale di Lejeune, impossibile da verificare negli anni ’60, era che la presenza del cromosoma sovrannumerario comportasse un problema nel metabolismo cellulare con il risultato finale di intossicare i neuroni causando così la disabilità intellettiva. Un recente articolo pubblicato dalla rivista Scientific Reports - opera del Professor Pierluigi Strippoli dell’Università di Bologna - ha individuato per la prima volta nel sangue e nelle urine dei bambini con sindrome di Down un profilo metabolico caratteristico; in particolare le sostanze rilevate in concentrazioni anomale sembrerebbero esserlo in modo proporzionale al modello previsto dai genetisti in presenza di un cromosoma in più. Questi studi contribuiscono a una più profonda comprensione delle basi biologiche della condizione.
5.2. L'Evoluzione della Comprensione del Legame con l'Età Materna
Un prestigioso studio della Yeshiva University di New York dà un’ulteriore conferma scientifica della correlazione tra incidenza della sindrome di Down ed età della futura mamma, spiegando le ragioni genetiche di questo legame. Il fatto che le donne con età avanzata siano più a rischio di partorire un neonato con la sindrome di Down è piuttosto noto, ma non erano ancora state formulate dalla comunità scientifica ipotesi di spiegazioni di tale correlazione. Recentemente una ricerca della Yeshiva University di New York ha indagato la causa di questo legame, individuando nella “ricombinazione” dei geni, che con l’avanzare dell’età diventa più frequente e più ravvicinata in senso temporale. Lo studio ha preso in esame un campione di circa 4.200 famiglie con almeno due figli e ha analizzato nello specifico proprio il processo di “ricombinazione” dei cromosomi. Si è osservato che, se in tale processo si presenta un errore oppure rimane incompleto, ne derivano delle anomalie cromosomiche, che possono portare appunto alla sindrome di Down. Tali anomalie, come è noto, sono più frequenti con l’aumentare dell’età della madre, anche se ciò non significa che le future mamme più giovani siano immuni da questa possibilità. La ricerca americana rappresenta un passo in avanti notevole nella conoscenza dei meccanismi di funzionamento della meiosi (divisione cellulare), che è un elemento molto importante per affinare ricerca e diagnosi precoce.
5.3. Nuove Prospettive Diagnostiche e Terapeutiche
La ricerca scientifica continua a esplorare nuove strade per migliorare la vita delle persone con sindrome di Down. Questo include lo sviluppo di tecniche diagnostiche sempre più precise e meno invasive, come dimostrato dall'avanzamento dei test NIPT, e la ricerca di trattamenti mirati per le specifiche comorbidità mediche. L'obiettivo a lungo termine rimane la possibilità di intervenire sui meccanismi molecolari alla base della trisomia 21, aprendo la strada a future terapie che possano mitigare gli effetti della condizione o, in un orizzonte ancora più lontano, offrire soluzioni correttive. La diagnosi è suggerita da anomalie fisiche e sviluppo anomalo ed è confermata dal cariotipo e da altre analisi citogenetiche. La gestione dipende dalle cure di supporto e dal trattamento delle manifestazioni e anomalie specifiche della malattia associata.