La gestione della gravidanza in donne affette da beta-talassemia rappresenta una sfida clinica complessa, ma grazie al perfezionamento delle tecniche di fecondazione assistita, del management ostetrico, del monitoraggio fetale e neonatologico, è sempre più possibile che una donna affetta da beta-talassemia instauri e porti a termine una gravidanza. Questo è reso possibile dalla collaborazione di più specialisti, quali l'ematologo, il genetista e altri professionisti sanitari. Scopo di questo studio è stato quello di evidenziare come, nonostante l'elevato rischio ostetrico e neonatologico, diventi necessario un attento controllo della salute materna e delle condizioni fetali attraverso il monitoraggio di vari parametri. Abbiamo pensato di considerare le problematiche incontrate inerenti alla gestione della gravidanza nelle pazienti talassemiche, sottolineando il rapporto tra management della gravidanza ed outcome gestazionale e neonatologico.
Comprendere la Beta-Talassemia: Una Patologia Ereditaria del Sangue
La beta-talassemia, conosciuta anche come anemia mediterranea o morbo di Cooley, è una grave patologia ereditaria a carico dei globuli rossi. È dovuta a un'alterazione del gene che sintetizza le catene beta, componenti dell'emoglobina, la proteina contenuta nei globuli rossi responsabile del trasporto dell'ossigeno nel sangue. L'emoglobina normale dell'adulto (Hb A) è formata da 2 coppie di catene chiamate alfa e beta. Il normale sangue adulto contiene anche ≤ 2,5% di emoglobina A2 (composta da catene alfa e delta) e < 1,4% di emoglobina F (emoglobina fetale), che ha catene gamma al posto delle catene beta. La talassemia deriva da una sbilanciata sintesi dell'emoglobina, causata dalla ridotta produzione di almeno una catena di polipeptidi globinici (beta, alfa, gamma, delta). Le talassemie sono un gruppo di anemie emolitiche ereditarie microcitiche caratterizzate da difetto nella sintesi dell'emoglobina.
La beta-talassemia si manifesta in diverse forme, basate sull'ereditarietà dei geni difettosi:
- Eterozigote (talassemia minor o tratto talassemico): dovuta alla mutazione di un solo cromosoma, ereditato dalla madre o dal padre. Queste persone sono portatrici sane e solitamente asintomatiche, con un'anemia microcitica da lieve a moderata.
- Omozigote (talassemia major o anemia di Cooley): quando il difetto è a carico di entrambi i cromosomi. Questa è una malattia grave che si manifesta entro il primo e il secondo anno di vita con anemia grave e conseguente necessità di trasfusioni.
- Beta-talassemia intermedia: un quadro clinico variabile, intermedio tra la talassemia major e minor, causato dall'ereditarietà di 2 alleli beta-talassemici (beta+/beta0 o casi gravi di beta+/beta+).
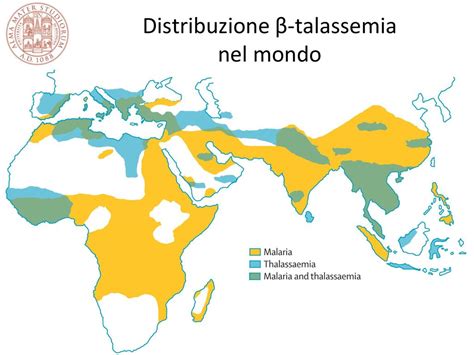
Geograficamente, la beta-talassemia è più frequente in soggetti originari del bacino del Mediterraneo, del Medio Oriente, del Sud-Est asiatico o dell'India. La malattia si concentra maggiormente in alcune aree dove la malaria era o è ancora endemica, colpendo a volte più del 20% della popolazione.
Le Sfide della Gravidanza in Donne con Beta-Talassemia
Le donne affette da beta-talassemia, specialmente nelle forme più severe come la talassemia major, affrontano una gravidanza considerata ad alto rischio ostetrico e neonatologico. Nonostante i progressi, la gestione richiede un approccio multidisciplinare e un monitoraggio costante. Una serie di problematiche devono essere considerate al riguardo, quali l'acquisizione di un adeguato consenso informato sui rischi materni e fetali, sui rischi riconducibili ad agenti infettivi, alla terapia con farmaci chelanti o antivirali, al rischio aggravato dallo stato di beta-talassemia per patologie cardiologiche ed endocrine.
Le complicanze endocrine rappresentano una sfida significativa. Il ritardo puberale e l'ipogonadismo continuano a essere le più comuni complicanze endocrine indotte dal sovraccarico di ferro, secondario alle ripetute trasfusioni, soprattutto nei pazienti non ben chelati o con scarsa compliance alla terapia chelante. Le cellule gonadotrope ipofisarie sono particolarmente sensibili all'accumulo di ferro, con conseguente ipogonadismo ipogonadotropo e ritardo di sviluppo puberale. Oltre al sovraccarico di ferro e al conseguente danno tissutale, altri fattori giocano un ruolo: l'anemia cronica con conseguente ipossia tissutale, l'epatopatia secondaria al sovraccarico di ferro o legata a epatiti croniche virali, i fattori nutrizionali, la carenza di zinco e le complicanze endocrine associate all'ipogonadismo. Oltre il 50% delle pazienti con TM presenta amenorrea primaria o secondaria a ipogonadismo ipogonadotropo (HH), particolarmente in caso di insufficiente ferro-chelazione; le rimanenti possono andare incontro, nel tempo, ad amenorrea secondaria, per alterata funzionalità dell’asse ipotalamo-ipofisi-gonadi.
Nei maschi, l'ipogonadismo si definisce con l'assenza dell'aumento del volume testicolare (meno di 4 ml). L'arresto della pubertà è una complicanza relativamente frequente nei pazienti con TM che hanno un accumulo di ferro moderato/grave ed è caratterizzato dall’assenza di progressione puberale nell'arco di un anno o più. In queste condizioni, il volume testicolare si arresta a 6-8 ml.
La gravidanza, pur non alterando la storia naturale della talassemia, può peggiorare lo stato clinico materno se non gestita adeguatamente. Il rischio principale per la madre è rappresentato dalle complicanze cardiache, che possono essere limitate assicurando una funzione cardiaca ottimale prima della gravidanza. Se la funzione cardiaca peggiora durante la gravidanza, la desferrioxanina (DFO) può essere somministrata con cautela, poiché la teratogenicità della DFO è dubbia. Riguardo ai ferrochelanti orali, mancano dati definitivi sulla fetotossicità. L'anemia cronica, se non adeguatamente gestita con trasfusioni, può esacerbare i rischi legati all'ipossia tissutale.
Diagnosi Prenatale Precoce: L'Innovazione della Celocentesi
Il progresso nella diagnosi prenatale ha rivoluzionato la gestione delle coppie a rischio di trasmettere patologie genetiche ai propri figli. Per la beta-talassemia, una delle innovazioni più significative è rappresentata dalla celocentesi. Questa tecnica, messa a punto dal ginecologo greco George Makrydimas e dalla sua équipe, con il contributo di Aurelio Maggio, direttore dell'Unità Operativa Ematologia II dell'Ospedale Vincenzo Cervello di Palermo, che ha sviluppato un metodo per isolare e analizzare le cellule embrionali raccolte, permette di ottenere una diagnosi certa con lo stesso valore predittivo dell'amniocentesi o della villocentesi, ma in una fase gestazionale molto più precoce.
La celocentesi consente di prelevare una piccola quantità di liquido celomatico (circa 1 ml) per via transvaginale, sotto guida ecografica, tra la 7a e la 9a settimana di gestazione. In questo periodo, la cavità celomatica, che ospita il sacco vitellino destinato a scomparire, contiene cellule di origine fetale. L'esame è definito invasivo, ma non comporta la perforazione della parete addominale come prevedono il prelievo dei villi coriali o del liquido amniotico. Le donne sottoposte a questa tecnica e che hanno sperimentato anche la villocentesi sostengono che il fastidio percepito con la celocentesi è decisamente minore. È la precocità dell'esame ciò che lo rende vantaggioso rispetto a villo e amniocentesi.
“Se il test conferma la patologia, i genitori possono decidere se interrompere la gravidanza a poche settimane dal suo avvio, invece di ricorrere a un aborto terapeutico in epoca più tarda, sicuramente più traumatico per la donna”, spiega Maggio. “Inoltre, in prospettiva futura, la possibilità di diagnosticare la talassemia così precocemente apre la strada alla fattibilità di interventi terapeutici in utero: la trasfusione di cellule staminali sane nell'embrione per curarlo della sua malattia.” Il rischio di interruzione spontanea della gravidanza legato al prelievo del liquido celomatico è inferiore all'1%, paragonabile a quello della villocentesi.
La tecnica di analisi messa a punto da Aurelio Maggio e colleghi è finalizzata alla diagnosi della beta-talassemia, ma sono già in corso studi per applicare la celocentesi alla diagnosi di altre patologie genetiche, come la fibrosi cistica. Con la pubblicazione sul British Journal of Haematology dei risultati dello studio condotto da Aurelio Maggio e George Makrydimas, la celocentesi ha superato lo stadio di tecnica sperimentale per diventare a tutti gli effetti un esame disponibile nella pratica clinica. La Fondazione "Cutino" svolge un importante ruolo di supporto logistico ed economico, accogliendo i familiari e supportando la ricerca. Circa il 30% della letteratura scientifica internazionale sulla celocentesi proviene dal gruppo di ricerca italiano.
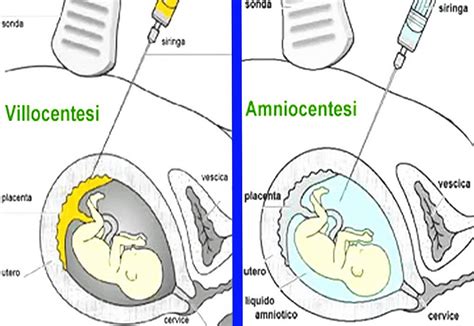
Altre metodiche di diagnosi prenatale invasive tradizionali includono la villocentesi (solitamente tra la 10a e la 12a settimana di gestazione) e l'amniocentesi (generalmente tra la 15a e la 20a settimana). Sebbene efficaci, queste procedure comportano un rischio di perdita fetale (aborto) pari all'1% per la villocentesi e l'amniocentesi, e al 2% per la cordocentesi, se effettuate in centri con reale esperienza. La celocentesi, offrendo una tempistica diagnostica notevolmente anticipata (7-9 settimane), permette decisioni più rapide e un approccio meno traumatico alla gestione della gravidanza.
Gestione Clinica della Gravidanza: Un Approccio Multidisciplinare
Una volta confermata la gravidanza in una donna con beta-talassemia, la gestione clinica deve essere affidata a un'équipe multidisciplinare composta da ostetrico, ematologo ed endocrinologo. La paziente deve essere informata che, sebbene la gravidanza sia ad alto rischio, l'esito è di solito favorevole con un appropriato monitoraggio.
Monitoraggio Materno e Fetale:Il monitoraggio di vari parametri è essenziale. Include la valutazione della salute materna, con particolare attenzione alle condizioni cardiache ed endocrine, e il monitoraggio delle condizioni fetali. L'obiettivo è mantenere la concentrazione di emoglobina pre-trasfusionale intorno a 10 g/dL durante la gravidanza. La ferritina sierica, indicatore del carico di ferro, aumenta in genere del 10% durante la gestazione.
Terapia Trasfusionale e Chelatrice:Le trasfusioni di globuli rossi sono spesso necessarie per mantenere adeguati livelli di emoglobina e prevenire l'anemia grave, che può portare a ipossia tissutale. L'incremento trasfusionale durante la gravidanza mira a mantenere l'emoglobina a circa 9-10 g/dL. La terapia chelante del ferro è fondamentale per rimuovere il ferro in eccesso accumulato a causa delle trasfusioni frequenti e del maggiore assorbimento dovuto all'eritropoiesi inefficace. La terapia chelante è generalmente iniziata quando i livelli di ferritina sierica superano i 1000 ng/mL o dopo circa 1-2 anni di trasfusioni programmate. Per quanto riguarda i farmaci chelanti orali, mancano dati definitivi sulla loro fetotossicità, mentre la desferrioxanina (DFO) può essere somministrata con cautela in caso di peggioramento della funzione cardiaca. Dopo il parto, la DFO può essere ripresa, ma non gli altri chelanti orali.
Gestione Endocrina e Riproduttiva:Le donne con talassemia possono portare a termine una gravidanza, ma la decisione di concepire dovrebbe essere presa con attenzione insieme al talassemologo. Le ragazze con TM che presentano amenorrea primaria o secondaria necessitano di specifico trattamento ormonale per stimolare la produzione ovarica e indurre l'ovulazione. Le opzioni per la fertilità dipendono dallo stato del partner e dall'entità del danno dell'asse ipotalamo-ipofisi-gonadi. L'induzione dell'ovulazione dovrebbe essere effettuata solo da specialisti della medicina riproduttiva.
Fecondazione Assistita: il ruolo dell'endometrio e della riserva ovarica
Consulenza Pre-Gravidanza:Prima di iniziare qualsiasi trattamento per la fertilità, è cruciale che le pazienti e i loro partner effettuino una consulenza pre-gravidanza. Questa ha tre scopi principali:
- Valutazione dell'eleggibilità: Considerare attentamente la presenza di insufficienza cardiaca (valutata tramite Risonanza Magnetica con gradiente T2* per quantificare il ferro cardiaco), disfunzione epatica (tramite test biochimici e RM) e rischio di trasmissione verticale di virus (screening per HIV, Epatite B, Epatite C e Rosolia; vaccinazione anti-rosolia). Le pazienti HCV-RNA positive dovrebbero effettuare un trattamento antivirale prima della gravidanza. Si effettuano anche screening per diabete, funzione tiroidea e anticorpi acquisiti anti GR.
- Revisione dei trattamenti in corso: Rivalutare la terapia in atto, fornire consigli su dieta, fumo, alcol e iniziare la supplementazione con acido folico, calcio e vitamina D. La ferrochelazione deve essere sospesa. La terapia ormonale sostitutiva va sospesa almeno 4-6 settimane prima dell’induzione della gametogenesi. I bifosfonati sono controindicati durante la gravidanza e l'allattamento. Le pazienti ipotiroidee in trattamento con tiroxina devono incrementare le dosi.
- Discussione dei rischi: Dialogo tra medici, pazienti e partner riguardo ai rischi associati alla fertilità indotta e alla gravidanza.
Aspetti del Parto e Post-Parto
Per quanto riguarda il tipo di parto, se la gravidanza non è complicata, si può procedere mediante parto spontaneo. Tuttavia, circa l'80% delle donne con talassemia necessita del parto cesareo a causa dell'alta incidenza di sproporzione cefalo-pelvica, dovuta alla bassa statura e all'alterazione scheletrica delle pazienti, associata a crescita fetale normale.
Dopo il parto, la DFO può essere ripresa, ma non gli altri chelanti orali. L'allattamento al seno deve essere incoraggiato in tutti i casi, tranne in quelli in cui la madre è HCV-RNA positiva e/o HBsAg positiva, a causa del rischio di trasmissione dell'infezione. Supplementi con calcio e vitamina D devono essere somministrati durante l'allattamento, mentre la terapia con bifosfonati per l'osteoporosi deve essere ripresa alla fine dell'allattamento.
La Fertilità Maschile e le Nuove Frontiere Terapeutiche
Nei maschi con talassemia major, l'ipogonadismo e l'arresto puberale sono complicanze frequenti. L'induzione della spermatogenesi è più difficile rispetto all'induzione dell'ovulazione nelle femmine, con percentuali di successo variabili. Tuttavia, le nuove tecniche di micromanipolazione come l'iniezione di sperma intracitoplasmatico (ICSI) hanno aumentato la percentuale di concepimenti, anche in pazienti con oligo e astenospermia. Pertanto, lo sperma dovrebbe essere cryopreservato in tutti i soggetti, eccetto in caso di azoospermia, per preservare meglio la fertilità. La recente letteratura sui danni del DNA spermatico nei pazienti con talassemia accresce dubbi circa i rischi mutageni, specialmente dopo ICSI.
Nonostante la presenza di ipogonadismo, nella maggior parte dei pazienti con TM la funzione gonadica è di solito integra, pertanto la fertilità, soprattutto nelle femmine, è mantenuta. L'ovulazione nelle femmine e la spermatogenesi nei maschi possono essere indotte dalla terapia con gonadotropine esogene, bypassando l'asse ipotalamo-ipofisi. Altri disordini endocrini, come diabete e ipotiroidismo, possono influenzare il risultato del trattamento della fertilità e necessitano di terapia appropriata. È essenziale pianificare la gravidanza, sia spontanea che indotta, poiché queste gravidanze sono ad alto rischio per la madre e per il bambino. Tuttavia, questi rischi possono essere ridotti con la consulenza pre-gravidanza con l'ematologo, lo specialista della medicina riproduttiva, il cardiologo e l'ostetrico.
Prognosi e Prospettive Future
L'aspettativa di vita è normale per i soggetti con beta-talassemia minor o alfa-talassemia minor. La prognosi dell'emoglobinopatia H e della beta-talassemia intermedia varia. L'aspettativa di vita è ridotta nei soggetti con beta-talassemia major, principalmente dovuta a complicazioni da trasfusioni croniche. Tuttavia, i progressi nella cura della talassemia major attraverso la terapia trasfusionale e ferrochelante ottimale hanno determinato un aumento della sopravvivenza dei pazienti fino all'età adulta, migliorando notevolmente la qualità di vita e aumentando le aspettative di avere una famiglia.
La diagnosi e la consulenza genetica prenatale sono pratiche standard in caso di feti a rischio e possono guidare la terapia fetale. Le talassemie più gravi vanno sospettate in pazienti con anamnesi familiare positiva, sintomatologia indicativa o anemia emolitica microcitica. Qualora sia sospettata una talassemia, vanno eseguiti i test di laboratorio per anemia microcitica ed emolitica e l'analisi quantitativa dell'emoglobina.
Il trattamento per le forme gravi prevede trasfusioni, splenectomia, terapia chelante e trapianto di cellule staminali. Il trapianto allogenico di cellule staminali è l'unica opzione curativa e deve essere considerato in tutti i pazienti idonei. Nuove terapie come il luspatercept, una proteina di fusione ricombinante iniettabile che inibisce la segnalazione della via del fattore di crescita trasformante-beta, rappresentano un'opzione di trattamento nei pazienti dipendenti da trasfusione, riducendo il fabbisogno trasfusionale. In prospettiva futura, la ricerca sulla terapia genica offre la speranza di poter curare la talassemia alla radice, correggendo il difetto genetico alla base della malattia.
La gestione clinica della gravidanza in donne con beta-talassemia è un esempio emblematico di come l'avanzamento della medicina e la collaborazione tra diverse specialità possano trasformare patologie potenzialmente letali in condizioni gestibili, consentendo a molte donne di realizzare il desiderio di maternità con esiti favorevoli per sé e per i propri figli.
tags: #donna #affetta #da #talassemia #partorisce #in