La fibrosi cistica (FC) è una patologia multiorgano estremamente complessa, una malattia cronica e progressiva che si manifesta nelle prime tappe dell’infanzia e che colpisce numerosi organi del corpo, come i polmoni, l’intestino, il pancreas, il fegato. È una malattia genetica ereditaria che si trasmette in forma recessiva, interessando in ugual modo maschi e femmine. Ogni persona possiede due copie del gene, ognuna delle quali proviene da entrambi i progenitori. Se una persona dispone di una sola mutazione della coppia del gene, viene definita portatrice della malattia ma non malata. La sua incidenza nella popolazione italiana è di circa 1 individuo su 2.500 affetto, mentre i portatori sani costituiscono una percentuale molto più ampia, circa 1 individuo su 25.
La fibrosi cistica è causata da varianti patogene di un gene specifico chiamato Cftr (cystic fibrosis transmembrane conductance regulator). Le mutazioni in queste aree codificanti possono modificare la composizione della proteina CFTR e inattivarla, portando a conseguenze significative sulla salute.
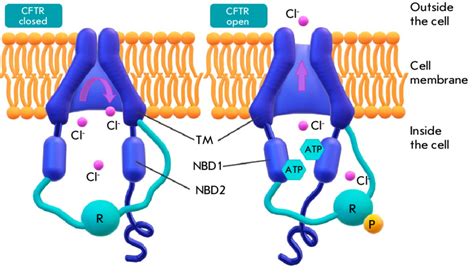
Meccanismi Patologici e Manifestazioni Cliniche
Nei polmoni dei pazienti affetti da fibrosi cistica, la mutazione della proteina CFTR è associata alla disidratazione del liquido superficiale, generando muco spesso e denso. Questo muco denso ostruisce le vie aeree, rendendole più soggette a infezioni croniche e infiammazioni, e in alcuni casi può causare insufficienza respiratoria.
Il sistema digerente è altrettanto compromesso. Il muco denso impedisce anche agli enzimi digestivi del pancreas di raggiungere l’intestino tenue, il che porta a notevoli difficoltà nel digerire i grassi e nell’assorbire alcuni nutrienti essenziali. Questo può avere ripercussioni sulla crescita e sullo stato nutrizionale generale del paziente. Alcuni malati di FC contraggono anche malattie al fegato, aggiungendo un ulteriore livello di complessità alla gestione clinica.
La malattia si può manifestare in oltre duemila varianti geniche che danno origine ad almeno sei classi sintomatiche diverse. Data la difficoltà di studiare e conoscere tutte le forme in cui può manifestarsi, tutte le terapie finora sviluppate agiscono prevalentemente sui sintomi e non sono risolutive in senso assoluto. L’ereditarietà recessiva autosomica implica che un bambino affetto da FC eredita una copia del gene difettoso da ciascun genitore.
Diagnosi Precoce: Test Neonatali e Test del Sudore
Indagare precocemente la presenza della fibrosi cistica è fondamentale per iniziare tempestivamente le cure e migliorare significativamente la qualità di vita dei piccoli pazienti. Oggi, la diagnosi precoce si avvale di due strumenti principali: lo screening neonatale e il test del sudore.
Screening Neonatale: Il Dosaggio della Tripsina
Lo screening neonatale consiste in un piccolo prelievo di sangue, effettuato dal tallone del neonato tra le prime 48-72 ore di vita. Questo esame si basa sul dosaggio della tripsina, un particolare enzima pancreatico. Se il test rileva livelli elevati di tripsina, ciò non indica automaticamente che il bambino sia affetto da FC. In questo caso, il Centro Regionale di Riferimento per gli Screening richiama la famiglia per effettuare un secondo dosaggio enzimatico, generalmente entro il 15° o 20° giorno di vita del bambino. Questo esame è indolore e non invasivo, eseguibile in ambulatorio. Un intervento precoce, se le cure vengono iniziate sin dai primi mesi di vita, può migliorare la crescita, aiutare a mantenere sani i polmoni, ridurre i ricoveri ospedalieri e prospettare una migliore qualità di vita. Se lo screening è negativo, le famiglie non ricevono alcuna comunicazione.
Il Test del Sudore: Misurazione del Cloro
Il test del sudore è considerato il modo più efficace per diagnosticare la FC. Si tratta di un semplice test indolore e non invasivo, eseguito in ambulatorio. Consiste nella misurazione della concentrazione di sale (cloro) nel sudore. Il limite al di sopra del quale il test è ritenuto positivo, confermando la diagnosi di FC, equivale a 60 mEq/L. Al contrario, risulta negativo con valori di cloro al di sotto di 40 mEq/L. Per i valori all’interno di questo range (tra 40 e 60 mEq/L), il test può essere ripetuto per fornire una diagnosi accertata di FC.
Questo esame viene richiesto in diverse circostanze: nei bambini con screening neonatale positivo per FC, nei bambini che abbiano presentato alla nascita ileo da meconio (un'ostruzione intestinale precoce), nei bambini con sintomi respiratori o gastrointestinali che inducano a sospettare una diagnosi di FC, e nei casi di pancreatite cronica o grave disidratazione, soprattutto durante i mesi estivi.
Analisi Genetica e Diagnosi Preimpianto
Nello studio genetico vengono analizzate tutte le regioni codificanti del gene CFTR, ovvero quelle che contengono le informazioni necessarie per la sintesi della proteina. I cambiamenti, o mutazioni, in queste aree possono alterare la composizione della proteina e inattivarla. Data l’alta frequenza di questa malattia, lo studio genetico della CFTR rientra nei pannelli di screening genetico raccomandati.
Per le coppie che desiderano pianificare una gravidanza o che affrontano problemi di fertilità legati alla FC, è fondamentale richiedere un consiglio medico e genetico. Lo specialista suggerirà di ampliare lo studio genetico a entrambi i membri della coppia per valutare il rischio di trasmettere la malattia.
La Diagnosi Genetica Preimpianto (DGP) rappresenta una tecnica di riproduzione assistita avanzata che permette di selezionare gli embrioni sani per la fibrosi cistica prima dell'impianto. Questo approccio offre un'ulteriore opzione per le coppie portatrici del gene difettoso che desiderano avere figli senza il rischio di trasmettere la patologia.
Diagnosi genetica preimpianto | Clinica PMA Roma e Madrid | New Fertility Group
Fertilità e Procreazione Assistita nella FC
Gli uomini affetti da fibrosi cistica possono soffrire di problemi di fertilità. Un'ampia percentuale di questi pazienti è azoospermica, ovvero, nel loro seme non sono presenti spermatozoi. Questo è dovuto all'assenza o all'ostruzione dei condotti deferenti, attraverso i quali gli spermatozoi circolano prima di essere espulsi durante l'eiaculazione. Quando i malati di fibrosi cistica desiderano ampliare la famiglia, devono necessariamente richiedere un consiglio medico e genetico.
Gestione della Malattia e Terapie Innovative
Sebbene ad oggi non esista una cura definitiva per la fibrosi cistica, la ricerca medica ha compiuto passi da gigante nel migliorare l'aspettativa e la qualità di vita dei pazienti. Se fino a poche decine di anni fa la mortalità era alta già nei primi anni di vita, attualmente l'aspettativa di vita mediana si attesta intorno ai 40 anni, grazie al miglioramento delle strategie terapeutiche.
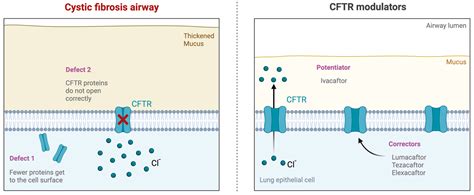
Oltre alle terapie di routine, per alcune mutazioni specifiche del gene CFTR, come ad esempio la F508del e altre meno frequenti, sono ora disponibili farmaci correttori e potenziatori. Questi farmaci innovativi intervengono direttamente sul funzionamento della proteina CFTR, migliorando il decorso della malattia. Negli ultimi anni, infatti, è stato approvato un nuovo farmaco, nato dalla combinazione di tre molecole diverse, che ha mostrato un'efficacia molto superiore, contribuendo a rivoluzionare la qualità di vita dei pazienti, specialmente quelli con le mutazioni più comuni del gene CFTR.
Uno dei problemi principali della malattia risiede nella variabilità delle mutazioni che la causano. Le varianti genetiche meno comuni sono state studiate in modo limitato e alcune non sono ancora state completamente caratterizzate, proprio per la difficoltà di studiarle in laboratorio e per la mancanza, fino a tempi recenti, di modelli di studio in vitro adeguati. Questo significa che almeno il 20% dei pazienti affetti potrebbe non disporre ancora di cure mirate approvate. L'aspettativa e la qualità di vita di questi pazienti subiranno un marcato miglioramento non appena queste varianti verranno studiate a fondo e verrà valutata la loro capacità di rispondere a farmaci più efficaci.
A tal fine, la ricerca sta esplorando nuove frontiere, come le tecniche di riprogrammazione cellulare. Studi recenti hanno utilizzato cellule staminali respiratorie prelevate dall'epitelio nasale dei pazienti con mutazioni rare di fibrosi cistica per farle crescere in coltura. Questo approccio consente di ottenere modelli di malattia cosiddetti "ex vivo", mediante speciali colture e la generazione di organoidi che riproducono in forma miniaturizzata e tridimensionale il tessuto respiratorio difettoso del paziente. Utilizzando questi modelli, è possibile valutare gli effetti delle specifiche mutazioni geniche sulle corrispondenti proteine difettose nelle cellule di ogni singolo paziente e testare l'efficacia di farmaci specifici nel correggere la proteina CFTR mutata, ripristinandone la funzionalità. Questo apre la strada a un approccio terapeutico sempre più personalizzato.
La gestione delle infezioni respiratorie è cruciale. Il muco vischioso che ostruisce le vie aeree favorisce la proliferazione batterica, rendendo l'antibioticoterapia (preferibilmente per via aerosolica) un elemento essenziale nel controllo delle infezioni. La prescrizione antibiotica dovrebbe basarsi sull'esito dell'esame microbiologico sulle secrezioni bronchiali. Inoltre, i pazienti necessitano di una dieta libera, equilibrata e ipercalorica per far fronte alle loro esigenze nutrizionali specifiche.
Il Sangue di Cordone Ombelicale: Potenziali Applicazioni e Donazione
Il sangue di cordone ombelicale, ovvero il sangue rimasto nella placenta e nel cordone dopo il parto, rappresenta una preziosa risorsa biologica. Le mamme e i papà possono decidere di donarlo, e questo sangue viene abitualmente utilizzato come sorgente di cellule staminali ematopoietiche, da impiegare per il trapianto di pazienti con malattie ematologiche che non dispongono di un donatore familiare compatibile.
Negli ultimi anni, la ricerca ha esplorato un potenziale utilizzo ancora più ampio per il sangue cordonale, in particolare per i neonati prematuri. I neonati di età gestazionale molto bassa, nati prima della 28esima settimana, necessitano spesso di ripetute trasfusioni di globuli rossi. Tradizionalmente, queste trasfusioni venivano effettuate con sangue donato dagli adulti. Tuttavia, il sangue degli adulti contiene un'emoglobina diversa dall'emoglobina fetale (HbF) presente nel neonato pretermine. L'emoglobina adulta tende a rilasciare una quantità maggiore di ossigeno ai tessuti, il che può provocare effetti tossici su organi delicati come la retina (retinopatia del prematuro), il tessuto cerebrale o il sistema respiratorio (displasia bronco-polmonare), poiché il sistema metabolico del bambino pretermine potrebbe non essere in grado di proteggersi adeguatamente da un potenziale danno ossidativo.
Studi pilota hanno dimostrato che le trasfusioni di globuli rossi ottenuti da cordone ombelicale aumentano l'emoglobina mantenendo elevati livelli di emoglobina fetale. Uno studio multicentrico italiano, denominato BORN (umBilical blOod to tRansfuse preterm Neonates), ha coinvolto diverse banche del cordone e unità di terapia intensiva neonatale. L'obiettivo era valutare la frequenza di retinopatia severa e l'impatto delle trasfusioni di sangue cordonale sulle altre patologie associate alla prematurità. Le analisi intermedie di sicurezza hanno dimostrato come le trasfusioni di sangue cordonale siano sicure e associate a un minor numero di eventi avversi rispetto a chi veniva trasfuso con sangue dell'adulto.
Per trasferire questo approccio dalla ricerca alla pratica clinica, è necessario espandere la pratica della donazione del sangue di cordone ombelicale. A livello normativo, si sta discutendo il riconoscimento dell'utilizzo del sangue da cordone anche a scopo trasfusionale, definendo requisiti di qualità specifici.
Donare sangue, o cordone ombelicale, è un gesto di generosità, civiltà e consapevolezza sociale. Sebbene la donazione di cellule staminali da midollo o sangue periferico sia più nota, è fondamentale che anche la donazione di cordone ombelicale riceva maggiore visibilità e informazione. La trasfusione di sangue cordonale può contribuire a divulgare una corretta informazione su questo tipo di donazione, a beneficio dei più piccoli pazienti che ne potrebbero trarre vantaggio. La raccolta di unità di sangue di cordone ombelicale donate alle banche pubbliche permette di preparare i concentrati di globuli rossi da utilizzare per le trasfusioni.
Considerazioni Legali e Risarcimento per Mancata Diagnosi Prenatale
La fibrosi cistica, essendo una grave malattia genetica, può dar luogo a questioni legali relative alla diagnosi prenatale. Avvocati specializzati in errori nella diagnosi prenatale e in mancata diagnosi di fibrosi cistica offrono assistenza in tutta l'Unione Europea. Le richieste di risarcimento possono sorgere in diverse circostanze:
- Gravidanze indesiderate a causa di un'errata diagnosi.
- Coppie che, progettando un figlio, si sottopongono a esami genetici che tuttavia non rilevano la malattia.
- Situazioni in cui il medico non propone di effettuare un test genetico sul feto o commette errori nell'eseguirlo.
Esistono esami che possono determinare in tempo utile se il feto sia affetto da fibrosi cistica, consentendo ai genitori di scegliere se interrompere o meno la gravidanza. Il danno ai familiari del bambino nato con una patologia congenita a causa di un'errata diagnosi prenatale dà diritto al risarcimento di tutti i danni conseguenti alla privazione del diritto di interrompere la gravidanza. In virtù della propagazione degli effetti protettivi del contratto stipulato dalla gestante con il ginecologo, il risarcimento può competere anche al padre e alle sorelle della minore.
Per i genitori il cui bambino è risultato affetto da fibrosi cistica sin dalla nascita, e l’ostetrico non ha offerto di eseguire un test genetico nelle prime fasi della gravidanza, o il test effettuato non ha diagnosticato la malattia, è possibile richiedere una consulenza legale. Studi legali con una vasta esperienza nel trattamento di casi di nascita indesiderata offrono le conoscenze giuridiche e l'esperienza necessarie per assistere le famiglie nell'ottenere l'assistenza finanziaria necessaria a ridurre o eliminare l'onere finanziario causato dalla malformazione genetica o dalla malattia ereditaria.
Diagnosi genetica preimpianto | Clinica PMA Roma e Madrid | New Fertility Group
tags: #cordone #ombelicale #fibrosi #cistica