La diagnosi prenatale rappresenta una pietra angolare nella medicina moderna, offrendo ai futuri genitori la possibilità di acquisire informazioni cruciali sulla salute del proprio bambino prima della nascita. Tra le tecniche disponibili, la villocentesi si distingue come un metodo invasivo di elevata precisione, indispensabile per l'analisi del DNA fetale e l'identificazione di una vasta gamma di malattie genetiche. Questo include la capacità di rilevare difetti genetici che possono predisporre a cardiopatie congenite, fornendo un quadro diagnostico fondamentale per la gestione della gravidanza e la pianificazione delle cure future.
La Villocentesi: Tecnica, Procedura e Significato
La villocentesi è una tecnica invasiva di diagnosi prenatale che consente di effettuare un prelievo di villi coriali dalla placenta inserendo un ago sotto guida ecografica attraverso l’addome materno. Questa procedura è cruciale per ottenere il materiale genetico necessario per un'analisi approfondita. I villi coriali sono il tessuto che costituisce la placenta e derivano dallo stesso uovo fecondato che si differenzia in embrione, placenta e membrane. Di conseguenza, il patrimonio genetico contenuto nelle cellule placentari è identico a quello dell’embrione e può essere utilizzato per gli stessi scopi diagnostici dell’amniocentesi, ma con il vantaggio di una maggiore precocità.
Quando e Come si Esegue la Villocentesi
Il prelievo di villi coriali può essere effettuato a partire dalla decima settimana compiuta di gravidanza in poi, solitamente a partire dalla 11° settimana di gestazione. In rari casi, la posizione della placenta può rendere possibile l’esecuzione dell’esame solo dopo le 12-13 settimane, come nel caso dell'utero retroversoflesso. Questa finestra temporale precoce rappresenta un vantaggio significativo rispetto ad altre tecniche diagnostiche, poiché fornisce risultati in una fase iniziale della gravidanza. Abitualmente, la procedura può trovare la sua applicazione fino al termine della gravidanza in situazioni cliniche specifiche.
Il giorno dell'esame di Villocentesi - Diagnosi prenatale, vengono effettuate diverse fasi preparatorie e l'intervento vero e proprio. Innanzitutto, è prevista un'ecografia del primo trimestre e un'ecografia ostetrica: il ginecologo ecografista effettuerà un’ecografia per la valutazione di eventuali anomalie fetali, per confermare l’epoca gestazionale, il numero dei feti, la vitalità e la morfologia di questi, la quantità di liquido amniotico e la localizzazione placentare. Occorre anche misurare le dimensioni del feto e scegliere la zona più adatta per l'introduzione dell'ago. Segue la consulenza genetica: il genetista medico valuterà la storia personale e familiare della coppia, determinando i possibili rischi e orientando le scelte diagnostiche. Il prelievo dei villi coriali viene eseguito sotto controllo ecografico, per via transaddominale. Questa tecnica prevede che il sottile ago sia spinto lungo la placenta, parallelamente alle membrane, per minimizzare il rischio di perforazioni, e quindi si aspirino i villi con un ripetuto movimento di va e vieni. Con la tecnica particolare adottata in centri specializzati, che vanta un'esperienza trentennale, l’esame è pressocché indolore e dura solamente pochi secondi, circa 20. Altri operatori, per ridurre le difficoltà tecniche, utilizzano la tecnica del doppio ago (si infigge prima un ago più grosso attraverso il quale se ne fa passare un altro più sottile) che, tuttavia, risulta più traumatica ed invasiva e non è necessaria con operatori esperti. Il materiale aspirato viene subito posto in una capsula sterile per valutarne la quantità e, qualora questa sia insufficiente, si procede ad una seconda aspirazione, evenienza rarissima.
Per quanto riguarda la preparazione della paziente, la mattina dell’esame non è previsto il digiuno. Tuttavia, in caso di assunzione di anticoagulanti (eparina, cardioaspirina, aspirinetta, ecc.), questi dovranno essere sospesi quattro giorni prima dell’esame e per i quattro giorni successivi, previa indicazione medica. Dopo il prelievo, la signora sarà accompagnata in sala privata per un riposo post-prelievo, e consigliamo di astenersi da attività fisiche importanti per almeno 2-3 giorni. È anche consigliabile assumere un antispastico dal giorno precedente l'esame per ridurre l’eventuale insorgenza di contrazioni uterine e rimanere a riposo a casa per 2-3 giorni. È sconsigliabile praticare l’esame in presenza di episodi febbrili della madre ed in caso di minaccia di aborto in corso. Nelle gestanti con gruppo sanguigno Rh negativo e con partner Rh positivo, è necessario effettuare, dopo la villocentesi, la profilassi anti-D mediante somministrazione di immunoglobuline specifiche al fine di ridurre il rischio di isoimmunizzazione Rh, una patologia che può avere conseguenze gravi per il feto o neonato. Nelle donne già immunizzate, l'esecuzione della biopsia dei villi coriali è controindicata.
Rischi e Sicurezza della Villocentesi
La villocentesi presenta, rispetto all’amniocentesi, il vantaggio della precocità diagnostica, dato che si può eseguire 3-4 settimane prima, e fornisce gli stessi risultati con un rischio di aborto simile o minore. Il rischio di aborto viene generalmente quantificato nello 0,5-1%, cioè un caso ogni 100-200 procedure. L’incidenza di aborto e complicanze è strettamente legata alla capacità e all’esperienza dell’operatore, e il rischio può tranquillamente essere ridotto o aumentato in modo significativo. In casistiche con operatori esperti, l’esperienza ha consentito di ridurre il rischio di aborto al di sotto dello 0,5% e di azzerare quasi le complicanze. Il prelievo dei villi coriali aumenta dell'1-3/1000 il rischio di perdita fetale (rispetto a quello naturale di qualsiasi gravidanza) e non è quindi differente da quello registrato dopo amniocentesi. È stato segnalato un aumento del rischio di parto pretermine. Le complicanze infettive sono rare, più frequenti dopo i prelievi transcervicali che richiedono più di un tentativo. Il rischio di lesioni fetali causate dall'ago è trascurabile, ove si consideri che il prelievo deve essere effettuato sotto controllo ecografico continuo. Tuttavia, è importante notare che il prelievo dei villi coriali eseguito prima della 10a settimana di gestazione può associarsi ad un aumento del rischio di provocare lesioni degli arti del feto, motivo per cui è fondamentale rispettare le tempistiche indicate.
Gestione delle Gravidanze Gemellari
Nelle gravidanze gemellari bisogna distinguere fra gravidanze mono e bicoriali. Nelle bicoriali, cioè quando vi sono due sacche e due placente diverse, è necessario avere un campione di entrambi i feti e quindi eseguire due diversi prelievi. Questo è dovuto al fatto che i gemelli bicoriali hanno ereditato dai genitori un corredo genetico differente, né più né meno come succede per i fratelli che non sono gemelli. Tra le diverse tecniche riportate in letteratura, la più diffusa e meno rischiosa consiste nell'effettuare due prelievi distinti e consecutivi, con introduzione di due aghi distinti l'uno dopo l'altro sotto controllo ecografico continuo. Al contrario, nelle gravidanze monocoriali-monoamniotiche, è sufficiente un unico campionamento, poiché i gemelli che hanno una sola placenta e che possono essere contenuti nello stesso sacco amniotico o in due sacchi differenti (gemelli monocoriali), sono geneticamente identici. Nelle gravidanze monocoriali si procederà al campionamento e al prelievo da un solo sacco amniotico, quando la diagnosi di monocorionicità sarà definitivamente accertata, in assenza di anomalie morfologiche di uno o entrambi i feti e quando la crescita fetale non sia discordante. Nel caso di anomalie ecografiche di uno o di entrambi i feti, in presenza di due sacchi amniotici differenti si procederà al prelievo in entrambi le sacchi, per escludere l'eventualità di mosaicismi.
Profondità Diagnostica: Dalle Anomalie Cromosomiche alle Malattie Monogeniche
La villocentesi non è un semplice screening, ma una vera e propria diagnosi. Attenzione, non confondere un esame di diagnosi prenatale (villocentesi o amniocentesi) con gli screening prenatali, che sono test statistici sul teorico rischio dell’esistenza della sola sindrome di down. Un tempo la si riteneva utile anche ad ipotizzare un rischio di cardiopatia. Mai farsi spaventare o rassicurare da un test di questo tipo, poiché si tratta di screening. L'analisi statistica è un esame per valutare il rischio, con più o meno accuratezza, esclusivamente della Sindrome di Down e Trisomia 18. Al contrario, la villocentesi fornisce una diagnosi certa di un ampio spettro di condizioni.
Screening in gravidanza e diagnosi prenatale - Dr.ssa Claudia Tironi
Analisi Cromosomica Tradizionale e Aneuploidie
L’esame dei villi coriali, quindi, viene effettuato per esaminare il cariotipo fetale, al fine di evidenziare la presenza di eventuali anomalie cromosomiche, o per la diagnosi di eventuali malattie genetiche. L’indicazione principale alla villocentesi è appunto rappresentata dallo studio dei cromosomi fetali. Questi sono presenti nelle cellule del nostro organismo nel numero di 46 o, per meglio dire, di 23 coppie di cromosomi omologhi che derivano in parti uguali dal padre e dalla madre. I cromosomi possono presentare delle anomalie di numero o struttura; vi possono essere quindi cromosomi in più o in meno (aneuploidia), oppure con anomalie della loro struttura. Il caso più frequente e conosciuto è la Sindrome di Down o Mongolismo, che è causato dalla presenza di 47 cromosomi con un 21 in eccesso; infatti, viene anche definita Trisomia 21. L’incidenza di anomalie dei cromosomi aumenta in parallelo con l’età materna, ma anche nelle donne giovani si possono avere casi di anomalie. Ad esempio, se la madre ha 20 anni le probabilità che nasca un bimbo Down sono di 1:1.105, se ha 30 anni 1:723, se ne ha 40 1:92. Il rischio complessivo per tutte le anomalie dei cromosomi è invece all’incirca il doppio. La diagnosi può essere fatta attraverso la coltura delle cellule dei villi e l’osservazione al microscopio dei cromosomi (tecnica tradizionale), processo che richiede tempi di attesa di circa 15-18 giorni.
Per le aneuploidie cromosomiche più comuni (cromosomi 13, 18, 21, X e Y), per sindrome di Down, Edwards, Patau, sesso e anomalie cromosomiche del sesso, si utilizza una tecnica molecolare avanzata. Il nostro Centro offre (inclusa nel prezzo) la possibilità di ottenere una risposta rapida mediante una tecnica molecolare avanzata di amplificazione genica (PCR), completamente automatizzata, conosciuta come Quantitative Fluorescent - Polimerase Chain Reaction (QF-PCR), che consente di ottenere i risultati in sole 24/48 ore. Questo esito preliminare, basato su una tecnica diversa (QF-PCR), prevede l’impiego di sonde a DNA specifiche per i diversi cromosomi ed ha una attendibilità del 95% circa.
Cariotipo Molecolare e Microalterazioni (Array-CGH)
Più recentemente, utilizzando una metodica di biologia molecolare, chiamata microarray-Comparative Genomic Hybridization (CMA o cariotipo molecolare), è possibile identificare anche alcune patologie derivanti da alterazioni submicroscopiche non visibili con la tecnica tradizionale. La recente introduzione della tecnica microarray CGH, basata sulla biologia molecolare, consente di effettuare un approfondimento diagnostico di secondo livello che si esegue per integrare l’esame tradizionale. Con questa tecnica non è necessaria la coltura cellulare, e quindi i tempi diagnostici sono più brevi (3-5 giorni), ciò consente una risposta più veloce anche in epoca gestazionale avanzata, e non vi è il rischio di fallimento della coltura cellulare. L’analisi con microarray consente di identificare, oltre alle tradizionali aneuploidie, alcune patologie derivanti da alterazioni cromosomiche submicroscopiche (ad esempio la Sindrome di DiGeorge, la cri-du-chat, la Prader-Willi, etc.) non evidenziabili con il cariotipo tradizionale. È particolarmente indicata in feti con malformazioni, Traslucenza Nucale molto elevata ed a supporto dello studio citogenetico tradizionale. La villocentesi molecolare, di più recente introduzione (metà degli anni ‘2000), va a ricercare microdelezioni e microduplicazioni mediante tecnica COMPARATIVE GENOMIC HYBRIDITATION (aCGH). Questa ricerca, da alcuni chiamata anche “cariotipo molecolare” per la possibilità di diagnosticare anche molte aneuploidie, non può essere disgiunta dalla citogenetica tradizionale, pena la possibilità di mancare diverse diagnosi legate a difetti cromosomici.
Diagnosi di Malattie Genetiche Specifiche (DNA Molecolare)
Tramite tecniche di biologia molecolare può inoltre essere eseguita l’analisi diretta di frammenti di DNA per la diagnosi di svariate malattie genetiche quali la talassemia, la fibrosi cistica, la Sindrome del Cromosoma X Fragile (ritardo mentale), la Sordità Congenita, la Distrofia Muscolare di Duchenne-Becker ed altre, i deficit metabolici. Queste malattie possono essere trasmesse da genitori portatori sani, totalmente inconsapevoli di esserlo.
Fibrosi Cistica (FC)
La fibrosi cistica (FC) è una grave malattia ereditaria che colpisce alla nascita 1 bambino su 2500. Nei pazienti affetti da FC, le secrezioni (cioè i liquidi biologici come il muco, il sudore, la saliva, lo sperma, i succhi gastrici) sono molto più dense e viscose del normale. I problemi più gravi sono a carico dei polmoni, dove il muco estremamente denso può causare problemi respiratori e infezioni. La FC è una malattia che si trasmette con modalità autosomica recessiva, determinata da alterazioni del DNA, chiamate “mutazioni”, che insorgono in entrambe le copie del gene CFTR (Cystic Fibrosis Transmembrane Regulator). Negli soggetti malati, entrambe le copie del gene sono alterate. Gli individui che possiedono una sola copia del gene alterato e una normale sono invece privi di ogni sintomo, ma sono portatori sani e avranno una probabilità del 25% di avere figli affetti da FC. Attualmente sono note circa 1500 mutazioni responsabili e con incidenza variabile. Oggi, attraverso un test genetico (studio del DNA), è possibile eseguire sul liquido amniotico o sui villi coriali la ricerca delle mutazioni più frequenti che causano la FC (screening 34 o 48 mutazioni), che nel complesso permette di identificare circa l’85% dei casi di FC nella nostra popolazione. Se l’analisi molecolare dovesse riscontrare una mutazione, permane comunque un rischio residuo di circa 1 su 500 che il feto sia affetto da FC, per la presenza di un’altra mutazione più rara. La mutazione 2183AA->G è una mutazione non rara (rappresenta il 9.3% delle mutazioni nel Nord-Italia), funzionalmente di tipo "frameshift" e quindi probabilmente appartenente alla classe I, perciò responsabile di un'alterazione importante della struttura della proteina CFTR. Di certo si può dire che è una mutazione che, quando associata ad un'altra mutazione "severe", comporta una forma di FC "classica", con insufficienza pancreatica e danno polmonare. L'entità della malattia polmonare è difficile da prevedere nel singolo caso. Con molte riserve vanno tenuti presenti i risultati forniti da alcune ricerche su piccoli numeri (3 con genotipo 2183AA->G /2183AA->G, 9 con genotipo 2183AA->G /altra mutazione), da cui risulterebbe mutazione "severa" anche a livello polmonare.
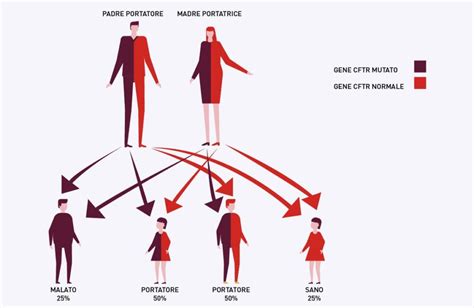
Un esempio pratico di come la diagnosi prenatale si inserisce nella vita delle coppie è il caso di una coppia di portatori di FC. In tale situazione, hanno avuto una figlia sana di dieci anni, ma successivamente una figlia è morta appena nata perché affetta da FC. Dopo questa tragica perdita, la coppia ha intrapreso quattro tentativi di avere altri figli sani, avviando quattro gravidanze, tutte sottoposte a indagine genetica e tutte diagnosticate con presenza di malattia FC e quindi interrotte. La diagnosi prenatale, in questi casi, è stata eseguita attraverso prelievo di villo e analisi genetica. L'accuratezza di questo tipo di diagnosi prenatale è estremamente elevata (può essere considerata del 99%). In parole semplici, la possibilità di una risposta sbagliata è bassissima (meno dell'1%), purché siano soddisfatte alcune condizioni: le mutazioni da ricercare nel villo siano conosciute perché sono quelle di cui i genitori sono portatori, il prelievo di villo sia eseguito da mani esperte, e il laboratorio abbia ampia consuetudine con l'indagine genetica che deve eseguire. Soddisfatte le condizioni di partenza, non ci sono dubbi sui risultati della procedura, che viene eseguita in ogni gravidanza in maniera del tutto indipendente dai precedenti risultati. La causa di tanta sfortuna è un'altra: è che ad ogni gravidanza una coppia di portatori ha sempre un rischio del 25% di avere un figlio malato. Questo rischio non cambia a seconda del numero di gravidanze e del loro esito. Questo rischio si è tradotto in una diagnosi di FC cinque volte in successione. Una frase che i genetisti usano è che "il caso non ha memoria": il caso può portare a ripetuti risultati sfavorevoli, come in questa coppia, come invece potrebbe portare alla nascita di figli tutti sani in un'altra coppia che fosse composta sempre da due portatori della stessa mutazione. Questa coppia deve essere informata che per evitare l'esperienza di altri aborti, potrebbe ricorrere alla fecondazione assistita e alla diagnosi genetica preimpianto (rivolgendosi ad un centro fuori d'Italia perché in Italia attualmente la legge 40/2004 vieta alle coppie di portatori FC la fecondazione assistita e quindi la diagnosi genetica preimpianto). Le ricerche svolte indicano che una delle più frequenti motivazioni delle coppie che ricorrono alla diagnosi genetica preimpianto è proprio una storia di aborti ripetuti per cause genetiche e il desiderio di evitarli.
Sindrome del Cromosoma X-Fragile (Ritardo Mentale)
L’X-Fragile è una malattia ereditaria causata dall’alterazione di un gene (FMR1) localizzato nel cromosoma X, e colpisce molto più frequentemente i maschi rispetto alle femmine, dato che queste ultime possiedono 2 copie del cromosoma X. Lo sviluppo mentale delle persone affette da tale malattia è molto vario. È noto che nella maggior parte dei casi di X-Fragile, l’alterazione responsabile della sindrome è l’espansione, attraverso le generazioni, di un tratto di DNA del gene FMR1, costituito da una sequenza ripetuta di tre basi nucleotidiche (CGG). Mentre nelle persone normali queste basi sono ripetute in un numero variabile da 5 a 58 volte, nelle persone malate sono invece ripetute più di 200 volte. Alcune persone possiedono un numero di ripetizioni intermedie all’interno del gene FMR1 (da 59 a 200) che non provocano alcun effetto, e sono definiti pazienti premutati. L’esame rileva feti di sesso maschile affetti dalla patologia, feti di sesso maschile e femminile premutati (portatori), e rari casi di sesso femminile affetti.
Distrofia Muscolare di Duchenne (DMD)
La DMD è determinata da alterazioni di un gene localizzato nel cromosoma X, che contiene le informazioni per la produzione di una proteina chiamata distrofina. Le mutazioni possono essere di vario tipo e comprendono sia delezioni (le più frequenti), sia sostituzioni nucleotidiche, ma tutte hanno come effetto quello di causare l’assenza totale della proteina. Come tutte le malattie legate al cromosoma X, la DMD si manifesta solo nei maschi (che hanno un solo cromosoma X), mentre le femmine, a parte alcune eccezioni, sono portatrici sane (perché possiedono un altro cromosoma X, oltre a quello mutato, che può compensarne le funzioni). Con il nostro studio genetico si analizza circa il 70% dei casi dovuti alle principali delezioni (18 esoni) e perdita di vaste regioni geniche.
Sordità Congenita
La sordità congenita è una malattia molto comune che colpisce, nella popolazione italiana, circa 6 milioni di persone. Il rischio di ricorrenza della Sordità Congenita per cause genetiche, ambientali o infettive alla nascita è di 1 caso su 1000. L’analisi molecolare prenatale del gene CX26 permette di ricercare nel DNA fetale la presenza delle mutazioni più frequenti che causano la malattia. Ad oggi sono state identificate ben 90 mutazioni del gene CX26. Il presente screening rileva esclusivamente le sordità da alterazione del gene GJB2 che codifica per la proteina connexina 26, le cui mutazioni sono responsabili di circa la metà dei casi di sordità ereditaria. Con lo screening non è possibile rilevare tutte le mutazioni del gene GJB2 ma, con la nostra metodica si giunge ad una accuratezza di circa l’ 80-90 % delle mutazioni.
Atrofia Muscolare Spinale (SMA)
La SMA rappresenta un gruppo di 4 forme di gravi disordini neuromuscolari, piuttosto conosciute per gli effetti devastanti che conducono alla progressiva degenerazione dei neuroni fino all’impossibilità di compiere movimenti. Si trasmette in modalità autosomica recessiva e si stima che in Italia vi sia 1 portatore sano e inconsapevole ogni 35-50 soggetti.
La Rivoluzione della Diagnosi Prenatale: Dalla Villocentesi Tradizionale alla NGPD
La diagnosi prenatale ha visto un'evoluzione significativa negli anni, passando da tecniche basilari a metodologie estremamente sofisticate che permettono di esplorare il genoma fetale con una profondità senza precedenti. Si configurano in genere due situazioni cliniche: quella routinaria, la cui scelta è legata alle decisioni della coppia e al loro più o meno profondo interesse a conoscere lo stato di salute del proprio figlio, e quella in cui gli accertamenti sono mirati alla presenza di una problematica specifica. Si tratta di accertamenti da eseguire nelle coppie “a rischio”. L’aspetto più interessante della diagnosi prenatale è quello legato al desiderio dei genitori di essere informati sul maggior numero di problematiche e con maggiore certezza.
Livelli di Indagine Diagnostica nella Villocentesi
Sulla base della comune esperienza è ormai noto che esistono diversi livelli di indagine sul liquido amniotico e sui villi coriali, ognuno con una capacità diagnostica crescente:
Villocentesi Tradizionale di Base: La villocentesi tradizionale, la prima ad essere introdotta all’inizio degli anni 70, è in grado di diagnosticare solo le patologie numeriche dei cromosomi (aneuploidie) e le maggiori alterazioni strutturali degli stessi. Indaga essenzialmente su quelle forme patologiche che interessano il numero e l’aspetto grossolano dei cromosomi. Nulla si potrà sapere delle piccole alterazioni dei cromosomi e sull’esistenza di alterazioni dei geni in essi contenuti. Essa svela solo il 5% dei portatori di anomalie genetiche.
Villocentesi con Studio Parziale del DNA: Introdotta a metà degli anni 90, descrive quanto si ricerca in aggiunta alla villocentesi tradizionale quando, attraverso l’impiego di metodiche genomiche diverse (es. MLPA o PCR ecc.), si ricercano le malattie genetiche più frequenti (fibrosi cistica, atrofia muscolare spinale (SMA), ritardo mentale da X fragile, sordità congenita ereditaria, distrofia muscolare di Duchenne). Questo gruppo di malattie genetiche aggiunge un 2% di soggetti diagnosticati, portando la capacità diagnostica complessiva al 7% delle patologie genetiche diagnosticate su 100 soggetti portatori di malattie genetiche.
Villocentesi Molecolare (aCGH) con Studio Parziale del DNA: La villocentesi molecolare, di più recente introduzione (metà degli anni ‘2000), va a ricercare microdelezioni e microduplicazioni mediante tecnica COMPARATIVE GENOMIC HYBRIDITATION (aCGH). Questa ricerca, da alcuni chiamata anche “cariotipo molecolare” per la possibilità di diagnosticare anche molte aneuploidie, non può essere disgiunta dalla citogenetica tradizionale, pena la possibilità di mancare diverse diagnosi legate a difetti cromosomici. La villocentesi molecolare con studio parziale del DNA (anch’essa introdotta a metà degli anni 2000) somma le precedenti. In molti laboratori è invalso l’uso di proporre una diagnosi più completa che comprendesse le informazioni aggiuntive sia del secondo che del terzo panel diagnostico. Aggiunge pertanto al 5% del cariotipo il 2% delle malattie genetiche più frequenti e l’1% delle microduplicazioni e microdelezioni, raggiungendo circa l'8% di patologie diagnosticate.
Next Generation Prenatal Diagnosis (NGPD) o Villocentesi Genomica: La vera rivoluzione diagnostica è avvenuta con l’attuale introduzione dei panel basati sullo studio dell’esoma (quella porzione del DNA che progetta il nostro organismo). Queste indagini hanno permesso di conoscere un numero teoricamente completo di patologie genetiche note. Tale panel è noto come villocentesi genomica ovvero TRIO o NGPD. In Italia è oggi possibile eseguire lo studio del genoma fetale. La villocentesi (Next Generation Prenatal Diagnosis o NGPD) fornisce tutte le informazioni oggi eticamente diagnosticabili con le più recenti metodologie genomiche. Questo tipo di ricerca arriva a diagnosticare tra il 70-80% delle malattie genetiche, una differenza impressionante rispetto all'amniocentesi o villocentesi tradizionale che svela solo il 5% dei portatori di anomalie genetiche. Non permette di giungere al 100% solo perché vengono escluse tutte quelle patologie estremamente rare, quelle ad origine genetica dubbia o sconosciuta oppure quelle per le quali non ci è “eticamente“ permesso di indagare. Benché si studino poche centinaia di geni (rispetto ai 19.000 astrattamente studiabili con le tecniche oggi in uso), questi rappresentano i geni che più frequentemente risultano alterati. La NGPD infatti non studia i polimorfismi di suscettibilità (SNP), cioè quelle varianti geniche che rendono l’uomo suscettibile ad un gran numero di malattie (quelle metaboliche come il diabete fino al cancro). Allo stesso modo la NGPD non prende in considerazione le malattie ad insorgenza tardiva, ad esempio l’Alzheimer, o quelle che coinvolgono l’aspetto psichiatrico del soggetto. Non studierà gli SNP, i cosiddetti polimorfismi di suscettibilità, cioè quelle varianti geniche che ci rendono suscettibili a qualsiasi malattie e soprattutto al cancro. Per semplificare, se in Italia tutte le donne eseguissero la NGPD, su circa 600.000 nuovi nati in un anno, solo poche decine di feti con malformazione genetica sfuggirebbero alla diagnosi. Le anomalie genetiche più frequenti verranno studiate ed escluse; le eventuali alterazioni individuate saranno valutate e illustrate in sede di consulenza genetica alla gestante. La residua possibilità di avere un figlio con problemi genetici resta legata a forme patologiche rarissime e a difetti multifattoriali, a volte non determinabili con certezza neanche dopo la nascita. Ovviamente si deve essere consapevoli che la natura potrebbe creare, occasionalmente, una nuova mutazione oggi non ancora conosciuta, e questo purtroppo non potrà essere diagnosticata come patologica fino a quando gli studi scientifici su questa nuova mutazione non abbiano stabilito una correlazione clinica precisa.
Applicazioni e Implicazioni per le Cardiopatie Genetiche
Le informazioni che fornisce la NGPD possono essere di guida per l’ostetrico, sia nella fase diagnostica della gravidanza che al momento del parto. Facciamo l’esempio di un feto che presenta, già dallo studio dei villi coriali, a 11 settimane, un difetto genetico per cardiopatia congenita. In questo caso il ginecologo seguirà con attenzione lo sviluppo del cuoricino onde studiare con maggiore attenzione il tipo e la gravità della malformazione cardiaca derivante dal messaggio genetico alterato, che come sappiamo, può esprimersi in misura diversa da caso a caso. L'entità della malattia polmonare, per esempio in caso di fibrosi cistica, è difficile da prevedere nel singolo caso. La probabilità di una tale associazione è particolarmente elevata in presenza malformazioni multiple (quadro polimalformativo). La NGPD, prevedendo lo studio del cariotipo fetale affiancato alle moderne tecniche di citogenetica molecolare (array-CGH) e di biologia molecolare (Next Generation Sequencing), consentirà di ridurre il rischio che il feto sia affetto da una specifica patologia genetica come nessun’altra indagine prenatale ha potuto e può fare. Identificare precocemente una predisposizione genetica a cardiopatie congenite permette un monitoraggio ecografico più mirato e l'organizzazione di un piano di assistenza specialistica sin dalla nascita.
Diagnosi Prenatale Molecolare Infettivologica
In caso di necessità diagnostica, la villocentesi Genetica può essere integrata dalla diagnostica prenatale molecolare infettivologica, che consiste nell’effettuare la ricerca con tecniche molecolari della presenza del genoma di agenti infettivi (es. Citomegalovirus, Herpes simplex, Varicella Zooster, Rubeovirus, HIV, Toxoplasma gondii, Parvovirus). Il vantaggio del ricorso alla tecnica molecolare (Polimerase Chain Reaction - PCR) risiede nel fatto che si ricerca direttamente il genoma, ossia la forma replicativa, dell’agente infettivo, superando i metodi tradizionali indiretti che esprimevano la produzione anticorpale fetale (IgM). Tali metodi infatti risultano molto imprecisi poiché dipendono molto dalla variabile maturità del sistema immunitario a sua volta legato all’età gestazionale. Non vi sono indicazioni ad effettuare uno screening infettivologico prima di eseguire la villocentesi. Nelle gestanti HIV positive non vi sono prove che la villocentesi aumenti il rischio di trasmissione del virus dalla mamma al feto, specie se il prelievo viene eseguito mentre è in atto una terapia adeguata con farmaci antiretrovirali.
Tempi di Risposta e Importanza della Consulenza Genetica
La tempistica per l’ottenimento dei risultati della villocentesi può variare a seconda del tipo di analisi richiesta. Come menzionato, gli esiti delle analisi rapide (sindrome di Down, sindrome di Edwards, sindrome di Patau), effettuate con QF-PCR, saranno disponibili entro 2 giorni, e saranno comunicati alla coppia direttamente dal genetista. Lo studio della mappa cromosomica fetale con tecnica tradizionale, che richiede la coltura cellulare, necessita di circa 15 giorni; la maggior parte di questo tempo serve per la coltura cellulare. È possibile talora che le cellule messe in coltura non crescano adeguatamente; si parla in questo caso di fallimento della coltura. Questa evenienza, estremamente rara e quantificabile all’incirca nello 0,5% dei casi, richiede un nuovo prelievo per allestire altre colture. Per le analisi che utilizzano l'array CGH, il tempo diagnostico è più breve, essendo una tecnica di biologia molecolare che non necessita di coltura cellulare, ed è possibile avere una risposta in 3-5 giorni, riducendo al minimo i tempi di attesa e l’ansietà della paziente. Per ricercare invece agenti infettivi, malattie genetiche o metaboliche specifiche, il tempo necessario per la diagnosi è compreso fra i 10 ed i 15 giorni. Tutte le altre indagini saranno disponibili entro 21 giorni.
In conclusione, ad oggi, la NGPD è la sola tecnica in grado di fornire in tempi strettissimi, e in maniera assoluta, la massima quantità di informazioni sullo stato di salute del feto. Le anomalie genetiche più frequenti verranno studiate ed escluse; le eventuali alterazioni individuate saranno valutate e illustrate in sede di consulenza genetica alla gestante. Il nostro Centro offre inoltre la possibilità di avere anche un esito preliminare nell’arco di 2 giorni. La sicurezza diagnostica dell’esame è molto elevata e gli errori sono assolutamente eccezionali se il genetista ha un’esperienza adeguata.
La consulenza genetica è un momento fondamentale in tutto il percorso della diagnosi prenatale. In sede di consulenza genetica saranno valutati i possibili rischi di coppia. Effettuiamo questa valutazione sulla base della storia personale e familiare della signora in gravidanza e del partner. Le coppie con gravidanze ad alto rischio di patologia saranno indirizzate verso analisi specifiche. Le coppie definite “a basso rischio” possono decidere se sottoporsi ad analisi di primo, secondo o terzo livello, questo sulla base del numero e della frequenza di patologie che intendono ricercare sul DNA fetale. Consigliamo di consultarsi con il proprio medico (e in famiglia) per decidere quali ricerche vuole eseguire sui villi coriali.
tags: #cardiopatie #genetiche #e #villocentesi