Nel complesso e delicato percorso della diagnosi prenatale, l'amniocentesi rappresenta uno strumento fondamentale per valutare la salute del feto. Tuttavia, la comprensione approfondita dei suoi risultati, in particolare quando si tratta di patologie genetiche complesse come la fibrosi cistica, richiede una chiara interpretazione che va oltre la semplice dicitura "negativa". Un'amniocentesi che non rileva mutazioni note per la fibrosi cistica, infatti, non sempre equivale a un'esclusione totale della malattia, introducendo il concetto cruciale di "rischio residuo" e la necessità di un'accurata valutazione clinica e genetica. Questa analisi esplorerà la fibrosi cistica, il ruolo dell'amniocentesi nella sua diagnosi, il significato profondo di un risultato negativo e le considerazioni essenziali che le future coppie genitoriali devono affrontare.
La Fibrosi Cistica: Una Malattia Ereditaria Complessa
La fibrosi cistica (FC) è una grave malattia ereditaria che colpisce alla nascita 1 bambino su 2500, posizionandosi come una delle patologie genetiche gravi più diffuse con cui un bambino può nascere. Si tratta di una condizione che si trasmette con modalità autosomica recessiva, il che significa che entrambi i genitori devono essere portatori sani di una mutazione genetica affinché il figlio possa sviluppare la malattia. Nel caso specifico della FC, essa è determinata da alterazioni del DNA, chiamate "mutazioni", che insorgono in entrambe le copie del gene CFTR (Cystic Fibrosis Transmembrane Regulator). Negli soggetti malati, entrambe le copie del gene sono alterate. Gli individui che possiedono una sola copia del gene alterato e una normale sono invece privi di ogni sintomo, ma sono portatori sani e avranno una probabilità del 25 % di avere figli affetti da FC ad ogni gravidanza. È un dato significativo che in Italia la statistica è di circa 1 portatore sano ogni 25 persone, evidenziando l'ampia diffusione di questa condizione genetica "silenziosa" nella popolazione.
Nei pazienti affetti da FC, le secrezioni, cioè i liquidi biologici come il muco, il sudore, la saliva, lo sperma e i succhi gastrici, sono molto più dense e viscose del normale. Questo fenomeno è dovuto alla totale assenza della proteina CFTR o al suo malfunzionamento, il quale interessa tutte le ghiandole a secrezione mucosa, in quanto vi è una carenza di acqua e di cloro. Di conseguenza, le secrezioni, essendo povere di acqua e di cloro, ristagnano e provocano un’ostruzione degli organi vitali come l'intestino, il pancreas e i bronchi. I problemi più gravi sono a carico dei polmoni, dove il muco estremamente denso può causare problemi respiratori e infezioni ricorrenti. Questa ostruzione persistente genera infezioni e infiammazioni che, con il passare del tempo, possono provocare una grave insufficienza respiratoria, portando a una progressiva perdita della funzionalità polmonare.
Accanto ai sintomi respiratori, si presenta una sintomatologia anche a carico del pancreas, che non riesce a svolgere la sua normale attività, ovvero quella di riversare gli enzimi all’interno dell’intestino. Questo disfunzionamento pancreatico porta a conseguenze come diarrea cronica con emissione di feci maleodoranti ed oleose, malassorbimento, difetti di digestione, ritardo di crescita nell’età infantile e scarso stato nutrizionale nell’età adulta. Il danno pancreatico con il passare degli anni può anche portare allo sviluppo di diabete di tipo 2. Altre manifestazioni della fibrosi cistica si presentano a carico dell’intestino, con possibili ostruzioni intestinali alla nascita (ileo da meconio) e spesso ostruzioni intestinali ripetute in età adolescenziale e adulta, delle cavità nasali, del fegato e, negli uomini, anche danni ai dotti deferenti che possono causare infertilità. Le tipiche manifestazioni della patologia includono difficoltà digestive in particolare dei grassi, delle proteine e degli amidi, e carenza di vitamine liposolubili. I principali sintomi osservabili sono affanno, respiro sibilante, infezioni polmonari e bronchiali frequenti, tosse persistente dapprima stizzosa poi catarrale, scarso accrescimento in peso e altezza e, infine, un caratteristico sudore salato. È importante sottolineare che la fibrosi cistica, in nessun modo, altera le capacità intellettive della persona e non altera l’aspetto fisico, tanto da essere spesso definita la “malattia invisibile”.
Le cure della fibrosi cistica, oggi, sono mirate ad alleviare i sintomi migliorando la vita del paziente e a prevenire le complicanze. I protocolli terapeutici, condivisi ed utilizzati a livello internazionale, sono adattati in base all’età e ai sintomi di ogni singola persona affetta dalla fibrosi cistica. La terapia è basata su antibiotici per le infezioni polmonari, farmaci fluidificanti, aerosol di antibiotici, fisioterapia respiratoria, nutrizione ipercalorica e trattamento delle complicanze. Nei casi più gravi, il trapianto polmonare è un intervento dedicato solo ai pazienti con insufficienza respiratoria irreversibile. Nonostante i progressi della ricerca, la fibrosi cistica rimane una malattia che riduce pesantemente la qualità e l’attesa di vita.
L'Amniocentesi: Uno Strumento Diagnostico Prenatale Fondamentale
L’amniocentesi è una procedura che consente il prelievo per via transaddominale del liquido amniotico al fine di effettuare una diagnosi prenatale di diverse condizioni, tra cui il cariotipo fetale, cioè le malattie cromosomiche del feto. Il periodo migliore per effettuare l’amniocentesi è tra la 16^ e la 18^ settimana di gravidanza, ovvero tra la fine del quarto e l'inizio del quinto mese. Per il prelievo si utilizza un ago sottile (21 gauge) che comporta solo un lieve fastidio per la paziente.
Esiste anche un'amniocentesi detta precocissima, praticata fra la 14a e la 16a settimana, che viene utilizzata soprattutto per la diagnosi di anomalie dei cromosomi. Con l'amniocentesi precocissima si preleva una minor quantità di liquido amniotico, proprio perché a quest'epoca di gravidanza ne è presente una minor quantità rispetto alle settimane successive. Di conseguenza, si ottengono un minor numero di amniociti, le cellule di origine fetale presenti nel liquido amniotico, che possono essere analizzati solo con tecniche speciali, come la CHG, FISH e QF-PCR, non ancora diffusamente disponibili e offerte solo in Centri altamente specializzati. Nel terzo trimestre, fra la 32a e la 39a settimana di gravidanza, l'amniocentesi può essere utilizzata per un'indicazione diagnostica differente, ovvero per valutare la maturazione dei polmoni del feto nelle donne in cui si consideri di anticipare il parto prima del termine della gravidanza.
L’amniocentesi è consigliata in diverse situazioni cliniche: alle donne che abbiano avuto un risultato positivo al test di screening prenatale (test combinato, bi test, tri test); alle donne che abbiano avuto figli con malattie cromosomiche o difetti del tubo neurale, come la spina bifida; alle donne con una età superiore ai 35 anni che non si siano sottoposte ai test di screening del primo trimestre; in caso di familiari (storia familiare) con specifiche malattie genetiche; e infine, in caso di risultati dell'ecografia che facciano sospettare la presenza di anomalie fetali. A prescindere dalle indicazioni mediche, la donna e il partner sono liberi di decidere se effettuare o non effettuare il test, dopo averne parlato a fondo con il medico o un sanitario esperto, come un medico genetista, il quale è spesso presente nei centri specializzati per un colloquio preliminare.
I cromosomi sono esaminati con tecniche avanzate come la Quantitative Fluorescent - Polimerase Chain Reaction (QF-PCR), che consente di ottenere i risultati sulle aneuploidie cromosomiche più comuni (cromosomi 13, 18, 21, X e Y), per sindrome di Down, Edwards, Patau, sesso e anomalie cromosomiche del sesso in sole 24/48 ore. Questa tecnica molecolare avanzata di amplificazione genica è completamente automatizzata e offre una risposta rapida. I risultati dell'amniocentesi consentono, nel 99% dei casi, di escludere o accertare (diagnosticare) numerose malattie genetiche, anche se non possono identificarle tutte.
In caso di necessità diagnostica, l’amniocentesi genetica può essere integrata dalla diagnostica prenatale molecolare infettivologica. Questa consiste nell’effettuare la ricerca con tecniche molecolari, specificamente la Polimerase Chain Reaction (PCR), della presenza del genoma di agenti infettivi, quali Citomegalovirus, Herpes simplex, Varicella Zooster, Rubeovirus, HIV, Toxoplasma gondii, Parvovirus. Il vantaggio del ricorso alla tecnica molecolare risiede nel fatto che si ricerca direttamente il genoma, ossia la forma replicativa, dell’agente infettivo, superando i metodi tradizionali indiretti che esprimevano la produzione anticorpale fetale (IgM). Tali metodi tradizionali, infatti, risultano molto imprecisi poiché dipendono molto dalla variabile maturità del sistema immunitario, a sua volta legato all’età gestazionale del feto.
Amniocentesi
Amniocentesi e Diagnosi di Fibrosi Cistica: Il Contesto della Ricerca
È possibile effettuare con l'amniocentesi la diagnosi di fibrosi cistica. L'analisi genetica consiste nella ricerca delle più comuni mutazioni del gene e viene effettuata sul DNA degli amniociti, che sono le cellule d'origine fetale presenti nel liquido amniotico. Oggi, attraverso un test genetico (studio del DNA), è possibile eseguire sul liquido amniotico o sui villi coriali la ricerca delle mutazioni più frequenti che causano la FC, utilizzando screening che analizzano 34 o 48 mutazioni. Nel complesso, questo tipo di screening permette di identificare circa l’85% dei casi di FC nella nostra popolazione.
È fondamentale, però, una precisazione cruciale: l'analisi genetica a cui il feto è sottoposto può dare un risultato sicuro solo nel caso in cui i genitori sappiano di essere entrambi portatori sani e le loro mutazioni genetiche siano conosciute. In tali circostanze, ci si accerta innanzitutto che il feto non abbia ricevuto da entrambi i genitori la mutazione CFTR da essi portata. Solo allora si potrà diagnosticare con elevato grado di certezza se il feto le ha ereditate, fornendo un quadro diagnostico molto affidabile.
Viceversa, se i genitori sono coppie della popolazione generale e non hanno effettuato analisi per la FC in precedenza, il test eseguito direttamente sul feto dà risultati incerti in una discreta percentuale di casi. In questi scenari, l'analisi genetica viene fatta in genere testando le mutazioni più comuni del gene CFTR. Questo test non copre tutte le mutazioni possibili ma ne copre dall'80 al 90% a seconda della regione di origine dei genitori. Di conseguenza, non si avrà un'esclusione con certezza assoluta della fibrosi cistica. Per esempio, se un'analisi molecolare dovesse riscontrare una mutazione, permane comunque un rischio residuo di circa 1 su 500 che il feto sia affetto da FC, a causa della possibile presenza di un'altra mutazione più rara che non è stata inclusa nello screening.
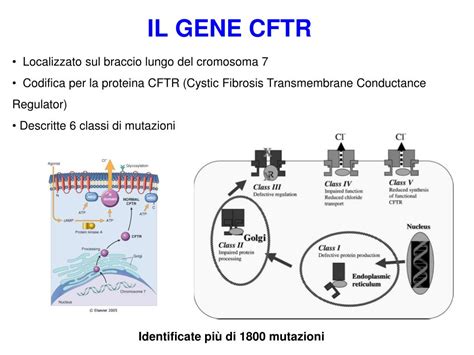
Il gene CFTR può essere difettoso in molti modi diversi: ad oggi, infatti, sono state scoperte più di 2.000 mutazioni; di queste, alcune sono frequenti e presenti in un numero elevato di portatori, molte altre sono rare o rarissime. Esistono varie tecniche di genetica molecolare per identificare le mutazioni del gene CFTR: i test più semplici, detti di 1° livello, identificano le mutazioni più frequenti, mentre i test più complessi, di 2° e 3° livello, anche quelle più rare. Nella popolazione italiana, oggi, un test di 1° livello è in grado di diagnosticare in media circa l’85% delle mutazioni che possono essere presenti sul gene CFTR, quindi l’85% dei portatori. Questa variabilità nella copertura del test sottolinea la necessità di una consulenza genetica approfondita per comprendere appieno le implicazioni di un risultato.
Il Significato di un Risultato "Negativo" per la Fibrosi Cistica
Un risultato di "normalità" o "negativo" per le mutazioni del gene della fibrosi cistica in seguito ad amniocentesi può sembrare rassicurante a prima vista, ma la sua interpretazione richiede una profonda comprensione delle limitazioni intrinseche dei test genetici e della genetica della malattia stessa. Quando un test prenatale sulla fibrosi cistica tramite amniocentesi indica che non sono state riscontrate le mutazioni più comuni, è essenziale comprendere che questa assenza non equivale a un'esclusione totale e assoluta della patologia.
Per una coppia di genitori appartenenti alla popolazione generale, che non hanno mai fatto il test del portatore e non hanno parentela con malati di fibrosi cistica, un'amniocentesi che non rileva mutazioni comuni del gene CFTR significa che il rischio generico di avere un figlio malato, che è di 1/2500 ad ogni gravidanza, si riduce significativamente ma non si annulla. Questo perché, come detto, il test delle mutazioni più comuni copre dall'80 al 90% delle mutazioni, a seconda della regione di origine dei genitori e del pannello di screening utilizzato. Pertanto, un risultato negativo non esclude con certezza assoluta la fibrosi cistica, poiché il feto potrebbe essere portatore di una mutazione più rara o addirittura sconosciuta, non inclusa nel pannello di screening. Questo è ciò che viene definito "rischio residuo".
Il concetto di rischio residuo è ulteriormente complicato dalla possibilità che il feto abbia ereditato una mutazione da un genitore e una seconda, più rara, dall'altro, anche se il test ha identificato solo la prima. Come accennato, se l’analisi molecolare dovesse riscontrare una mutazione, permane comunque un rischio residuo di circa 1 su 500 che il feto sia affetto da FC, per la presenza di un’altra mutazione più rara non rilevata. Questo scenario evidenzia che un singolo risultato positivo per una mutazione non è necessariamente sufficiente per una diagnosi completa e che la complessità del gene CFTR, con le sue oltre 2.000 mutazioni scoperte, rende la copertura totale una sfida diagnostica.
Esistono altri test più complessi per la ricerca delle mutazioni del gene CFTR, che identificano un numero maggiore di mutazioni. Si può arrivare a identificare circa il 90%, e difficilmente oltre, delle mutazioni note, e quindi questi test sono in grado di fornire una diagnosi più accurata. Tuttavia, essi non offrono mai l'esclusione completa del rischio di malattia. Inoltre, questi test più approfonditi richiedono tempi lunghi per l'esecuzione, il che può essere in contrasto con la necessità di avere chiarezza in tempi brevi durante una gravidanza. Un altro potenziale svantaggio è che questi test possono identificare "varianti" del gene il cui significato è oggi sconosciuto: non si sa se siano cioè vere mutazioni, capaci di dare malattia, o varianti innocue. Questo potrebbe ulteriormente complicare il problema, generando ansia e incertezza senza fornire risposte cliniche definitive.
Pertanto, la decisione di approfondire le indagini, magari con test di 2° o 3° livello, va molto valutata in concerto con il medico genetista. È essenziale considerare il profilo di rischio della coppia, la storia familiare e le implicazioni psicologiche di un'indagine che potrebbe non fornire risposte conclusive. La paziente che ha effettuato l’amniocentesi con risultato di normalità per le mutazioni del gene della fibrosi cistica, ma che appartiene a una coppia della popolazione generale non testata, dovrebbe essere pienamente consapevole che, sebbene il rischio sia notevolmente ridotto per le mutazioni più frequenti, una probabilità, seppur bassa, che il feto possa avere la fibrosi cistica, non può essere del tutto esclusa. Comprendere questo "rischio residuo" è il vero significato di un'amniocentesi "negativa" per la fibrosi cistica, un risultato che invita alla consapevolezza piuttosto che a una falsa sicurezza assoluta.
La Prevenzione e la Valutazione del Rischio: Il Test del Portatore Sano
Il test del portatore sano è un'indagine genetica cruciale eseguita su persone adulte sane che hanno intenzione di intraprendere una gravidanza e desiderano sapere se sono portatori di mutazioni del gene della fibrosi cistica e se corrono il rischio di avere figli con questa malattia. Considerando che la fibrosi cistica è la più comune tra le malattie genetiche gravi con cui un bambino può nascere e che lo stato di portatore sano è frequente (si stima 1 su 30 in Italia), tutte le coppie della popolazione generale che pianificano una gravidanza possono informarsi e decidere di fare il test.
Questo test ricerca nel corredo genetico, fatto di una lunghissima sequenza di DNA, la presenza di mutazioni a carico del gene CFTR: Cystic Fibrosis Transmembrane Conductance Regulator, il gene che, se difettoso (mutato), è responsabile della malattia. Come già menzionato, il gene CFTR può essere difettoso in molti modi diversi, con oltre 2.000 mutazioni scoperte ad oggi, alcune frequenti e molte altre rare o rarissime. I test di 1° livello, i più semplici e diffusi, sono in grado di diagnosticare in media circa l’85% delle mutazioni che possono essere presenti sul gene CFTR nella popolazione italiana, identificando quindi l’85% dei portatori.
In base a queste considerazioni, si può dire che chi risulta portatore di una mutazione ha un risultato certo (è sicuramente portatore della mutazione identificata), e quindi in questo caso il risultato è definitivo. Al contrario, chi risulta non portatore ha una probabilità bassa o bassissima, ma non del tutto esclusa, di esserlo, perché potrebbe avere una mutazione rara o sconosciuta. Questo è il concetto di “rischio residuo” e dipende da quanto sia stato approfondito il test, quindi da quante mutazioni rare o sconosciute potrebbe non aver identificato.
Il test del portatore sano è particolarmente raccomandato per le coppie che hanno parenti con fibrosi cistica o che sono già a conoscenza di essere portatori sani di mutazioni nel gene della fibrosi cistica. In questi casi specifici, il test è spesso fornito gratuitamente (o con costo ridotto) dal Servizio Sanitario Nazionale. Per le coppie della popolazione generale, invece, il costo del test non è generalmente coperto dal Servizio Sanitario Nazionale, a eccezione di alcune regioni come il Veneto, dove possono essere in vigore norme regionali particolari.
C’è un dibattito scientifico e sanitario sul fatto di inserire o meno il test per il portatore di fibrosi cistica tra quelli da suggerire alle coppie della popolazione “generale” in fase preconcezionale. A favore di questa proposta vi è il fatto che la fibrosi cistica è la più frequente delle malattie genetiche gravi con cui un bambino può nascere e resta ancora oggi, nonostante i progressi della ricerca, una malattia che riduce pesantemente la qualità e l’attesa di vita. Inoltre, la probabilità di essere portatori sani è elevata (1 su 30) e una coppia di portatori identificati dal test, e quindi con un rischio di 1 su 4 di avere ad ogni gravidanza un figlio malato, può utilizzare la villocentesi per avere una risposta certa, oltre che per altre patologie (es. Sindrome di Down), anche per la presenza o assenza di fibrosi cistica nel feto.
Le ragioni critiche a questa proposta includono il fatto che il test è basato su tecniche di genetica molecolare per lo più costose, non identifica tutti i portatori (la percentuale di “falsi negativi” varia a seconda della tecnica molecolare usata), può essere eseguito solo in laboratori altamente qualificati, e dà risposte che vanno commentate solo da personale specializzato. Per le coppie che non hanno figli malati e sono interessate a sapere se hanno rischio di generare figli malati di FC, non dovrebbero aspettare l'amniocentesi per valutare tale rischio. Infatti, l'amniocentesi viene fatta in un periodo avanzato di gravidanza, e questo renderebbe estremamente difficoltosa ogni decisione. Queste coppie dovrebbero preferibilmente richiedere il test del portatore (per ciascun membro della coppia) prima del concepimento.
Nell'ambito del test genetico per il portatore sano di fibrosi cistica, la presenza di polimorfismi poliT e poliTG viene frequentemente riportata, sollevando molti dubbi. Si tratta di variazioni della sequenza del DNA del gene CFTR che non hanno nella maggior parte dei casi significato patologico. Tutti i geni sono fatti di una sequenza di DNA, il quale a sua volta è costituito da molecole chimiche (basi azotate) poste secondo uno specifico ordine e posizione: queste basi si chiamano Adenina (A), Guanina (G), Timina (T), Citosina (C). Una mutazione genetica è una variazione della sequenza del DNA rispetto a quella presente nella maggior parte della popolazione sana; questa variazione è conosciuta come responsabile di un danno al funzionamento del gene e quindi in grado di provocare una malattia. In particolare, esiste una determinata regione del gene CFTR in cui si trova una porzione di DNA soggetta a particolare variabilità in cui sono presenti polimorfismi. In questo tratto la sequenza del DNA è costituita da una serie ripetuta di basi Timina, indicata con la lettera T. Circa l’80% della popolazione generale possiede nel suo patrimonio genetico la variante 7T, il 15% la variante 9T, mentre il 5% la variante 5T. Oggi si ritiene che le varianti 7T e 9T siano innocenti, per cui nella grande maggioranza dei casi non destano preoccupazioni se segnalate nel test del portatore sano. Più complicata è la valutazione della presenza della variante 5T: in alcuni casi, ma non tutti, questo polimorfismo può avere un effetto simile a quello di una mutazione. Il polimorfismo TG, invece, è costituito da due basi, Timina e Guanina, che possono essere ripetute un numero variabile di volte e quindi avere lunghezza variabile (da 9 a 13 ripetizioni, eccezionalmente fino a 15). La loro interpretazione richiede l'expertise di un genetista.

Alternative e Approfondimenti nella Diagnosi Prenatale
Oltre all'amniocentesi, esistono altre modalità per indagare i cromosomi e i geni, che offrono diverse tempistiche e livelli di invasività. Tra queste, la villocentesi e il test prenatale non invasivo (NIPT) rivestono un ruolo significativo.
La Villocentesi:La villocentesi, o prelievo di villo coriale (un frammento della placenta), è un'altra procedura invasiva di diagnosi prenatale. I tempi per l’esecuzione di villocentesi e amniocentesi sono molto diversi, e questo ha varie implicazioni. La villocentesi viene generalmente eseguita intorno alla decima settimana di gravidanza, fra la 10a e la 12a settimana, quindi significativamente prima dell'amniocentesi, che si pratica dopo la sedicesima settimana. Questa maggiore precocità di risposta dell'indagine la rende la procedura d'elezione se si chiede la massima precocità, per esempio, perché il rischio che il feto sia affetto da una particolare patologia è elevato, come nel caso della coppia di due portatori sani del gene FC.
È opportuno eseguire villocentesi o amniocentesi per fibrosi cistica solo nella gravidanza di una coppia costituita da due persone che sanno già di essere portatori sani di una mutazione nel gene CFTR. Sul villo coriale o su cellule da liquido amniotico prelevate durante l’esame si cercheranno le stesse mutazioni del gene CFTR di cui sono portatori i due partner, e si potrà diagnosticare con elevato grado di certezza se il feto le ha ereditate. Il test prenatale attraverso villocentesi è l’indagine consigliata nella coppia in cui entrambi i partner sanno di essere portatori di una mutazione del gene CFTR, poiché il rischio di fibrosi cistica nella gravidanza di questa coppia è elevato: 25%, ovvero una probabilità su 4. Il risultato del test può richiedere tempi variabili, da pochi giorni a una settimana, ed è solitamente disponibile circa poco prima della dodicesima settimana. Caratteristiche, vantaggi e svantaggi di entrambe le procedure invasive vanno chiesti e discussi a fondo con il ginecologo o altro sanitario esperto.
Il Test Prenatale Non Invasivo (NIPT):Tra le indagini non invasive, ha un ruolo di rilievo il NIPT, o Non-Invasive Prenatal Test, che consiste in un prelievo di sangue della madre eseguito a partire dalla nona settimana di gravidanza. In questo prelievo viene analizzato il DNA del feto entrato nel circolo materno. Sul DNA fetale vengono svolte indagini che forniscono un risultato indicante la probabilità che sia affetto da una certa malattia. Tuttavia, è cruciale comprendere che il NIPT è uno screening e non una diagnosi definitiva: un rischio alto dovrà essere confermato attraverso indagini più invasive, come l’amniocentesi o la villocentesi. Sono offerti test NIPT anche per malattie genetiche, tra cui la ricerca di mutazioni del gene della fibrosi cistica, ma per questa malattia ad oggi non si conoscono ancora i dati di sensibilità, specificità e margini di errore su larga scala.
Distinzione tra Analisi Cromosomica e Analisi Genica:È importante distinguere tra le indagini che si usano per analizzare i cromosomi e quelle che si usano per i geni. Le anomalie dei cromosomi riguardano il loro numero, come la sindrome di Down (in cui sono presenti tre cromosomi 21 invece che due), o la loro struttura (perdita o aggiunta o duplicazione di frammenti di cromosoma). Per indagare i cromosomi occorre un esame chiamato cariotipo, in cui i cromosomi, che sono le particolari strutture in cui si ripiegano i filamenti di DNA, risultano visibili e fotografabili con un esame specifico. D'altra parte, per indagare i geni, che sono collocati in precise posizioni all’interno dei cromosomi e sono piccolissime molecole di DNA, sono necessarie tecniche diverse chiamate indagini di genetica molecolare, conosciute attraverso il nome del singolo gene o dei vari geni che esaminano. Alcune malattie presenti fin dalla nascita (congenite) possono essere legate ad anomalie dei cromosomi, mentre altre ad anomalie dei geni, come nel caso della fibrosi cistica.
Altre Malattie Genetiche Diagnosticabili Prenatalmente:Nel contesto dell'amniocentesi o della villocentesi, oltre alla fibrosi cistica, è possibile eseguire test aggiuntivi per un ampio spettro di patologie genetiche. Tra queste figurano la Distrofia Muscolare di Duchenne (DMD), la Sordità Congenita, la mutazione del gene FMR1 associata alla Sindrome da X-Fragile (Sindrome di Martin-Bell o del cromosoma X fragile), e l'Atrofia Muscolare Spinale (SMA).La DMD è determinata da alterazioni di un gene localizzato nel cromosoma X, che contiene le informazioni per la produzione di una proteina chiamata distrofina. Le mutazioni, che possono essere di vario tipo e comprendono sia delezioni (le più frequenti) sia sostituzioni nucleotidiche, hanno tutte come effetto quello di causare l’assenza totale della proteina. Come tutte le malattie legate al cromosoma X, la DMD si manifesta solo nei maschi (che hanno un solo cromosoma X), mentre le femmine, a parte alcune eccezioni, sono portatrici sane (perché possiedono un altro cromosoma X, oltre a quello mutato, che può compensarne le funzioni).La sordità congenita è una malattia molto comune che colpisce, nella popolazione italiana, circa 6 milioni di persone. L’analisi molecolare prenatale del gene CX26 permette di ricercare nel DNA fetale la presenza delle mutazioni più frequenti che causano la malattia; ad oggi sono state identificate ben 90 mutazioni di questo gene.La sindrome di Martin-Bell o del cromosoma X fragile è la forma più comune di ritardo mentale dopo la sindrome di Down, in quanto interessa 1:4000 maschi e 1:6000 femmine. L’X-Fragile è una malattia ereditaria causata dall’alterazione di un gene (FMR1) localizzato nel cromosoma X, e colpisce molto più frequentemente i maschi rispetto alle femmine, dato che queste ultime possiedono 2 copie del cromosoma X. Lo sviluppo mentale delle persone affette da tale malattia è molto vario. Nella maggior parte dei casi di X-Fragile, l’alterazione responsabile della sindrome è l’espansione, attraverso le generazioni, di un tratto di DNA del gene FMR1, costituito da una sequenza ripetuta di tre basi nucleotidiche (CGG). Mentre nelle persone normali queste basi sono ripetute in un numero variabile da 5 a 58 volte, nelle persone malate sono invece ripetute più di 200 volte. Alcune persone possiedono un numero di ripetizioni intermedie all’interno del gene FMR1 (da 59 a 200) che non provocano alcun effetto. La disponibilità di un medico genetista per un colloquio preliminare all'amniocentesi è fondamentale per orientare la paziente e la coppia verso i test più appropriati e per fornire una consulenza completa.
Aspetti Legali e Diritti del Paziente
La diagnosi prenatale, con le sue complessità e implicazioni, tocca anche importanti aspetti legali, in particolare in relazione al risarcimento danni in caso di errori o omissioni. La fibrosi cistica, essendo una delle malattie genetiche gravi maggiormente diffuse, è frequentemente al centro di tali questioni. La persona malata eredita un gene difettoso sia dalla madre che dal padre che sono portatori sani del gene mutato CFTR, spesso senza saperlo, con una statistica in Italia di circa 1 portatore sano ogni 25 persone. I portatori sani, ad ogni gravidanza, hanno la possibilità di avere un figlio malato ogni quattro bambini.
Il risarcimento in caso di diagnosi di fibrosi cistica per il nuovo nato può essere richiesto in specifiche circostanze. Un primo scenario riguarda i casi in cui vi è stata una mal interpretazione o esecuzione errata degli esami genetici per la fibrosi cistica. Se i risultati sono stati falsati o non letti correttamente, portando a una mancata diagnosi, la famiglia può richiedere un indennizzo. Un secondo caso si verifica se una coppia affetta da fibrosi cistica, o che è a conoscenza di essere portatrice, decide di avere la gravidanza con una tecnica di fecondazione assistita di tipo eterologa e i gameti maschili o femminili ricevuti sono portatori della malattia. Ciò significa che l’azienda che ha fornito i gameti non ha eseguito i test specifici di screening per il donatore o la donatrice, non rilevando il loro stato di portatore, e quindi viene meno al suo dovere di diligenza. Un terzo scenario si presenta quando una coppia è a conoscenza di essere a rischio, ad esempio per storia familiare o perché entrambi i partner sono portatori accertati della mutazione CFTR, ma il medico non richiede nessun test per la valutazione fetale, impedendo così alla coppia di prendere decisioni informate.
I danni da risarcire alla famiglia coinvolta sono rivolti in primis alla madre del bambino nato affetto da fibrosi cistica a causa dell’errata diagnosi prenatale. A lei spetta il risarcimento di tutti i danni conseguenti alla impossibilità di interrompere la gravidanza, qualora avesse desiderato farlo in presenza di una diagnosi positiva e certa. Il risarcimento compete anche al padre e agli altri figli della coppia, in quanto l'evento di un figlio affetto da una grave patologia genetica comporta un significativo impatto emotivo, economico e organizzativo sull'intero nucleo familiare. La coppia che ha maturato la scelta di interrompere la gravidanza in caso di diagnosi di fibrosi cistica, una volta avuto il risultato del test, può richiedere l’interruzione presso gli ospedali che praticano questo intervento, secondo la legge 22 maggio 1978, n.194, che regola la richiesta di interruzione volontaria di gravidanza. È fondamentale sapere che il termine fissato dalla legge per eseguirla è non oltre la dodicesima settimana di gravidanza, se non per gravi motivi di salute della madre o del feto accertati da certificato medico.
La complessità della diagnosi prenatale e le sue implicazioni legali evidenziano l'importanza di un'informazione completa e accurata, di una consulenza genetica scrupolosa e dell'accesso a test affidabili. La consapevolezza dei propri diritti e delle possibilità di tutela legale è un elemento essenziale per le famiglie che si trovano ad affrontare queste difficili scelte.
tags: #amniocentesi #negativa #fibrosi #cistica