L'emoglobina (Hb) rappresenta una componente fondamentale della fisiologia umana, essendo una proteina contenuta nei globuli rossi responsabile del trasporto dell'ossigeno ai tessuti dell'organismo. Questa molecola, di forma sferica con un diametro di circa 5,5 nm, possiede una struttura tetramerica, formata da quattro subunità, ognuna delle quali chiamata globina. Oltre al trasporto dell'ossigeno, l'emoglobina gioca un ruolo cruciale nel metabolismo cellulare: una volta ceduto l'ossigeno agli organi, l'emoglobina lega l'anidride carbonica, uno dei principali prodotti di scarto del metabolismo, facendosi carico del trasporto ai polmoni mediante il circolo venoso. La complessità del sistema emoglobinico umano risiede nella presenza di varianti fisiologiche e atipiche, che si succedono durante le diverse fasi dello sviluppo, a seconda dei geni che vengono attivati in quel determinato periodo di vita.
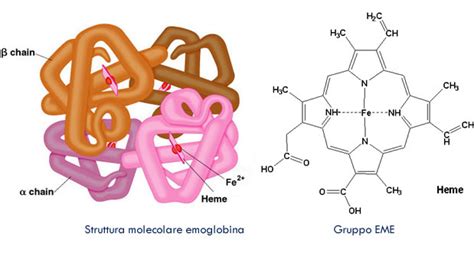
Struttura e proprietà dell'emoglobina fetale
Nei globuli rossi del feto è possibile identificare una forma di emoglobina diversa da quella adulta, nota come emoglobina fetale o HbF. Questa proteina è formata da due catene α (alfa) e da due catene γ (gamma), costituite rispettivamente da 141 e 146 amminoacidi. Mentre le due catene alfa sono identiche a quelle presenti nell'emoglobina adulta, le catene gamma differiscono dalle catene Beta (tipiche dell'adulto) per 39 amminoacidi.
Questa specifica modifica strutturale conferisce all'emoglobina fetale un'affinità per l'ossigeno superiore; in altre parole, si lega all'ossigeno in modo più tenace rispetto all'emoglobina adulta. Dal punto di vista funzionale, l'emoglobina fetale (HbF od emoglobina F) permette al feto di estrarre con maggiore efficacia l'ossigeno dal sangue materno. Il trasferimento di ossigeno al sangue fetale attraverso la barriera placentare è favorito anche dalla maggiore concentrazione di emoglobina, più alta di circa il 50% rispetto a quella del sangue materno. Si stima che l'HbF riesca a trasportare percentuali comprese tra il 20% e il 30% di ossigeno in più rispetto all'emoglobina materna, garantendo così la sopravvivenza e lo sviluppo del feto in un ambiente a bassa pressione parziale di ossigeno.
L'evoluzione delle emoglobine: dallo sviluppo all'età adulta
Durante lo sviluppo l’essere umano esprime diversi tipi di emoglobina. Esistono forme precoci, come le emoglobine Portland (ζ2γ2), prodotte nelle prime settimane dal concepimento. La sintesi delle globine Beta, caratterizzanti l'emoglobina adulta, è appena percettibile durante la vita fetale e raggiunge il normale regime soltanto verso la fine del terzo mese della vita extrauterina.
Nell'adulto sano, la composizione emoglobinica è dominata da:
- Emoglobina A1 (Hb-A1): Rappresenta il 97-98% del totale, costituita da due catene α e due catene β.
- Emoglobina A2 (Hb-A2): Costituisce il 2-3% del totale, formata da due catene α e due catene δ (delta).
- Emoglobina F (HbF): L'emoglobina predominante nella vita fetale, che diminuisce drasticamente dopo la nascita fino a percentuali dell'adulto, tipicamente entro il primo anno di vita, scendendo a livelli generalmente inferiori all'1%.
La differente espressione nel tempo, dal concepimento alla vita adulta, delle diverse catene globiniche nell'uomo dipende dall'attivazione e dallo spegnimento di specifici geni. Sebbene la struttura quaternaria cambi, queste emoglobine svolgono in linea generale la medesima funzione di trasporto, pur presentando affinità differenti per l'ossigeno.
Respirazione: il viaggio dell'ossigeno
Valori normali e determinazione clinica
In un adulto in salute, i livelli di emoglobina fetale considerabili normali oscillano tra lo 0.1% e l'1.1% (o, in alcuni range di riferimento, tra 0.3% e 1.2%). È fondamentale sottolineare che i valori di riferimento degli esami di laboratorio possono variare a seconda della metodologia di analisi dei campioni; pertanto, i dati indicati hanno uno scopo puramente informativo.
Per l’esame dell’emoglobina fetale viene richiesto un digiuno di almeno otto ore. Una piccola percentuale di emoglobina fetale viene espressa anche durante la vita adulta, e i suoi livelli possono variare in modo significativo sotto l'influenza di fattori quali l'età, il sesso o peculiarità genomiche. Quando negli adulti si riscontrano valori superiori all'1.1%, si parla di "emoglobina F alta". Tale riscontro, essendo una condizione perlopiù asintomatica, viene spesso rilevato casualmente in analisi del sangue effettuate per altri motivi. Una causa possibile di questo innalzamento è una condizione di recupero dovuta a ipoplasia di midollo osseo, ovvero un disturbo delle cellule staminali ematopoietiche.
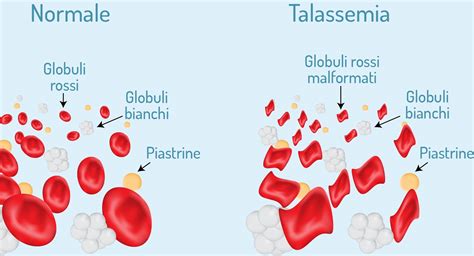
Significato patologico e talassemie
Il monitoraggio dell'emoglobina fetale è un pilastro nella diagnosi di diverse patologie ematiche ereditarie. Nell'utero, il feto normale produce una piccola quota di emoglobina adulta (2,5-5%). Al contrario, il feto affetto da talassemia maior ne produce una quantità ancor minore (inferiore al 2%). Per rilevare durante la gravidanza se un feto è affetto da talassemia maior, è possibile determinare la quantità di emoglobina adulta presente in un campione di sangue prelevato mediante cordocentesi.
Le talassemie sono causate da un difetto ereditario che impedisce la normale sintesi delle catene α (α-talassemia) o delle catene ß (ß-talassemia):
- α-talassemia: L'informazione genetica è contenuta in 4 geni. Il difetto può variare dall'alterazione di un solo gene (indistinguibile dalla normalità, ma trasmissibile) fino all'alterazione di tutti e quattro i geni (condizione non compatibile con lo sviluppo fetale). La delezione di tre geni porta alla formazione di complessi di quattro catene ß (HbH), associata ad anemia emolitica e splenomegalia.
- ß-talassemia: L'informazione è contenuta in 2 geni. L'alterazione di entrambi i geni (forma omozigote) definisce la ß-talassemia major o morbo di Cooley, caratterizzata da un'anemia molto grave che necessita di trasfusioni periodiche. L'alterazione di un solo gene (forma eterozigote), chiamata ß-talassemia minor o tratto talassemico, si presenta solitamente come una condizione asintomatica, caratterizzata da globuli rossi più numerosi ma di volume ridotto (microcitemia) e poveri di emoglobina.
Emoglobinopatie e persistenza ereditaria
Il termine "emoglobinopatia" racchiude circa 1000 tipi di disordini ematici ereditari, caratterizzati dalla presenza di varianti anomale dell'emoglobina o dalla riduzione della sua produzione. Circa il 7% della popolazione mondiale è portatore in eterozigosi di una variante genetica in una delle catene dell’emoglobina, con tassi di mutazione variabili in base all'etnia.
Tra le varianti più note figurano:
- Emoglobina S (HbS): Responsabile dell’anemia falciforme, che causa globuli rossi a forma di falce ed episodi dolorosi.
- Emoglobina C (HbC): Presente nel 2-3% delle persone di origine africana, con effetti clinici lievi.
- Emoglobina E (HbE): Comune nel sudest asiatico, associata a lieve anemia emolitica.
- Emoglobina di Bart: Si sviluppa nei feti affetti da alfa-talassemia, riflettendo una grave carenza delle catene alfa.
Esiste inoltre la "persistenza ereditaria dell'emoglobina fetale", una condizione benigna in cui concentrazioni importanti di emoglobina fetale (> 10%) persistono anche in età adulta. Questa peculiarità, solitamente asintomatica, è di grande interesse clinico poiché può alleviare la severità di certe emoglobinopatie e talassemie, fungendo da meccanismo di compensazione naturale.
Prospettive terapeutiche: il ruolo dell'idrossiurea
La ricerca medica ha dimostrato che una terapia farmacologica capace di aumentare la concentrazione di emoglobina fetale apporta benefici significativi ad alcune categorie di pazienti, in particolare quelli affetti da anemia falciforme o da talassemia Beta.
Il prototipo di questi farmaci è l'idrossiurea, un composto antineoplastico ad azione mielosopressiva. L'idrossiurea si è dimostrata efficace nell'aumentare i livelli di emoglobina fetale, riducendo conseguentemente l'incidenza di crisi dolorose nei pazienti affetti da anemia falciforme. Il trattamento delle emoglobinopatie può comportare anche l'utilizzo di terapie di supporto, mirate ad alleviare il dolore e minimizzare le complicanze. Nei casi di anemia grave, il ricorso a trasfusioni di sangue rimane, ancora oggi, una pratica clinica fondamentale per garantire l'apporto di ossigeno necessario ai tessuti, laddove la sintesi endogena sia compromessa o inefficiente.
tags: #vsg #emoglobina #fetale