I tumori renali nei bambini sono considerati rari, rappresentando circa il 5-6% di tutte le neoplasie diagnosticate al di sotto dei 15 anni di età. Tra queste forme tumorali, il più comune è il tumore di Wilms, noto anche come nefroblastoma, che costituisce circa il 90% dei tumori renali in età infantile. Questa neoplasia ha un'origine particolare, derivando da cellule embrionali del rene, e tende a svilupparsi prevalentemente nei primi anni di vita. La sua classificazione è stata riconosciuta per la prima volta come neoplasia renale da Rance nel lontano 1814 e successivamente descritta in dettaglio dal chirurgo Max Wilms nel 1899, nella sua monografia di 90 pagine intitolata “Die Mischgeschwülste der Niere”. Il nefroblastoma è un tumore embrionale, maligno e solido che si manifesta specificamente nel rene del bambino.
Che Cos'è il Nefroblastoma (Tumore di Wilms)?
Il nefroblastoma è il più comune tumore maligno del rene del bambino, caratterizzato da un'anomala proliferazione di cellule simili a quelle embrionali del rene, spesso definite come metanefroma. Questa peculiarità giustifica pienamente il termine di "tumore embrionale". La neoplasia trae origine dalle cellule renali precoci, denominate blastema nefrogenico, le quali normalmente si formano e si sviluppano durante la vita intrauterina, nel corso dello sviluppo embrionale. I reni, organi essenziali per filtrare il sangue ed eliminare le scorie tramite l'urina, oltre a regolare la pressione sanguigna e la produzione di globuli rossi, si formano a partire da queste cellule progenitrici. Può accadere che, dopo la nascita, alcune di queste cellule immature permangano all'interno dei reni. Se il DNA di tali cellule progenitrici subisce delle mutazioni, la cellula può acquisire nuove proprietà che la trasformano in una cellula tumorale. Questa, a sua volta, inizia a moltiplicarsi in modo incontrollato, dando origine al tumore di Wilms.
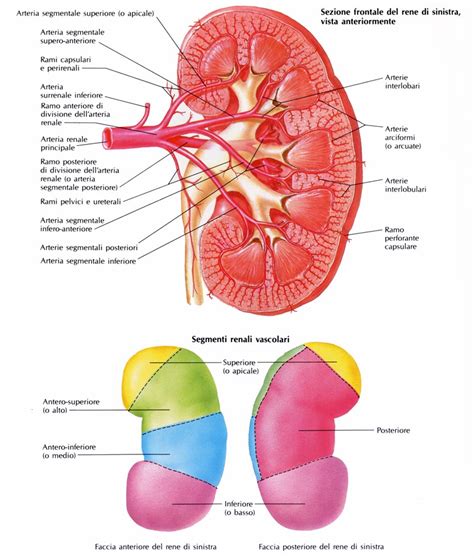
Il classico tumore di Wilms, spesso definito come "tipo misto", è un tumore renale complesso che presenta tre componenti istologiche principali: una componente blastemica, costituita da cellule immature; una componente epiteliale, che forma strutture simili a tubuli; e una componente mesenchimale, che costituisce lo stroma. La proporzione di queste tre componenti può variare notevolmente da caso a caso, il che significa che non sempre è possibile parlare di un tumore trifasico in senso stretto.
Epidemiologia e Incidenza: Un Tumore dell'Età Pediatrica
L'incidenza annuale del nefroblastoma si attesta intorno a 1 caso ogni 10.000 nati, e la malattia colpisce in maniera analoga i due sessi, sebbene alcune statistiche indichino una leggera maggiore incidenza nel sesso femminile. Questo tumore interessa soprattutto i bambini di età compresa tra 1 e 5 anni, con una maggiore frequenza di diagnosi intorno ai 3-4 anni di età. Tuttavia, il 15% di questi tumori viene diagnosticato in bambini con meno di un anno, e nel 2% dei casi in quelli con più di 8 anni. La diagnosi è meno frequente nei bambini di età superiore ai 10 anni ed è estremamente rara negli adolescenti e nei giovani adulti. In Europa e Nord America, il nefroblastoma colpisce circa 1 bambino su 10.000, con una prevalenza leggermente superiore nelle popolazioni africane e afroamericane e meno comune tra i bambini delle popolazioni dell'Asia orientale.
Nella maggior parte dei casi, il tumore di Wilms colpisce un solo rene, manifestandosi in forma monolaterale. Tuttavia, in una percentuale di casi che varia dal 5% al 10% (o circa il 7% secondo alcune fonti), possono essere interessati entrambi i reni, configurando una forma bilaterale della malattia.
Le Radici del Nefroblastoma: Genetica e Sindromi Predisponenti
Le cause precise del tumore di Wilms non sono ancora completamente note, ma si ritiene che fattori genetici giochino un ruolo significativo, soprattutto nelle forme associate a specifiche sindromi congenite. Nonostante ciò, nella stragrande maggioranza dei casi (circa il 99%), la malattia si presenta in modo sporadico, senza una chiara predisposizione ereditaria identificabile.
Il nefroblastoma si ritiene si sviluppi da piccoli ammassi di cellule renali anomale, noti come residui nefrogenici. Esistono diversi tipi di questi resti nefrogenici: alcuni possono rimanere dormienti per tutta la vita, mentre altri possono crescere in modo anomalo. Quando è presente un numero elevato di residui nefrogenici, la condizione viene definita nefroblastomatosi, che può essere diffusa o multifocale. La nefroblastomatosi è considerata una lesione potenziale precursore del nefroblastoma, e la sua definizione è in continua evoluzione, includendo tutte le lesioni che possono evolvere in questa neoplasia.
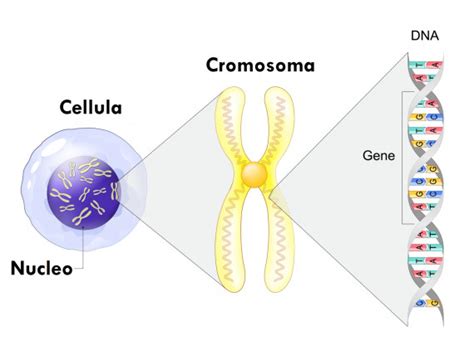
Nel contesto del nefroblastoma, sono state descritte varie anomalie a carico di diverse regioni cromosomiche. Tra queste, le più studiate includono le regioni 11p13 (che contiene il gene WT1), 11p15 (contenente il gene H19), 16q, 1p, 1q e 17p. Diversi geni svolgono un ruolo cruciale nello sviluppo del nefroblastoma. In particolare, il gene WT1, localizzato sul cromosoma 11p13, è un gene oncosoppressore, ovvero una porzione di DNA che funziona come un freno alla moltiplicazione cellulare. Questo gene interviene nelle prime fasi dello sviluppo embrionario del rene, influenzando la differenziazione dei nefroblasti. Delezioni a carico del gene WT1 sono state riscontrate nel 10-30% dei nefroblastomi. Oltre al WT1, il gene WT2, situato sul cromosoma 11p15.5, sembra avere un ruolo nello sviluppo del nefroblastoma. La perdita di eterozigosi (LOH), un meccanismo di sviluppo tumorale, è frequentemente riscontrata (circa il 40%) a livello del cromosoma 11p. Esistono anche indicazioni che suggeriscono l'esistenza di un terzo gene del tumore di Wilms, il gene WT3, la cui LOH si riscontra nel cromosoma 16q in circa il 20% dei tumori. Altri meccanismi di sviluppo del tumore includono mutazioni geniche e la perdita di imprinting (LOI).
Una percentuale minore, circa il 10% dei casi di nefroblastoma, si associa a difetti congeniti specifici, quali l'aniridia (una formazione incompleta dell'iride, la parte colorata dell'occhio), l'emi-ipertrofia (una condizione in cui una parte del corpo, come un braccio, è più sviluppata dell'omologa controlaterale) e diverse anomalie urogenitali. Il tumore di Wilms è inoltre più comune nei bambini affetti da alcune malattie genetiche rare che rientrano in quadri sindromici ben definiti. Tra queste troviamo la sindrome di Beckwith-Wiedemann, caratterizzata da un aumento delle dimensioni della lingua e di altre parti del corpo, oltre a una riduzione dei livelli ematici di glicemia. Un'altra è la sindrome di Denys-Drash, che si manifesta con anomalie genitali, nefropatia e la presenza del tumore di Wilms. La sindrome WAGR, invece, è caratterizzata dall'associazione di tumore di Wilms bilaterale con aniridia, anomalie urogenitali e ritardo mentale. Altre sindromi includono la sindrome di Perlman e la neurofibromatosi di Recklinghausen. Le forme familiari di nefroblastoma sono molto rare, interessando circa l'1% di tutti i bambini, e si trasmettono con un modello autosomico dominante a penetranza variabile. Tutti i tumori bilaterali rientrano in questa categoria ereditaria.
Manifestazioni Cliniche: Riconoscere i Segni
Il tumore di Wilms spesso si manifesta con sintomi poco specifici o, nelle fasi iniziali, può essere addirittura asintomatico. Il segno più comune e frequente è la comparsa di una massa addominale palpabile o di un gonfiore nell'addome, che spesso viene notato per la prima volta casualmente da un genitore o da un tutore durante il bagno o il cambio del bambino. Questa massa è solitamente non dolente, ma può causare una deformità visibile dell'addome.Raramente, i pazienti lamentano anche un dolore addominale, che si verifica in circa il 10% dei casi. Altri segnali che possono accompagnare la crescita del tumore includono ipertensione, febbre (riscontrata nel 20% dei casi), ematuria (presenza di sangue nelle urine), sebbene le tracce di sangue nelle urine non siano un segno frequente di questa forma tumorale, e anemia. In alcuni casi, possono presentarsi anche sintomi meno specifici come nausea, perdita di appetito, stitichezza e fiato corto.L'insorgenza dei sintomi può essere improvvisa, quasi da un giorno all'altro, con manifestazioni quali mal di pancia, febbre, perdita di peso e stanchezza. La tosse, in situazioni avanzate, può essere un sintomo clinico di metastasi polmonare. In rari casi, la manifestazione del tumore può essere repentina a causa di una rottura del tumore all'interno dell'addome, spesso in seguito a traumi accidentali, che provoca un'emorragia interna. È importante notare che diversi sintomi sono comuni ad altre malattie. Se i sintomi persistono, è fondamentale informare il pediatra per una valutazione tempestiva e accurata della causa.
Tumore di Wilms: guarire salvando la funzionalità renale
Il Percorso Diagnostico: Dagli Esami Strumentali all'Analisi Istologica
Se un bambino presenta sintomi che fanno sospettare un tumore di Wilms, il pediatra avvia un processo diagnostico approfondito. Questo inizia con l'anamnesi, durante la quale il medico si informa su quando e come si sono presentati i sintomi, sulla storia familiare del paziente e sulla sua storia clinica specifica. In particolare, si indaga sulla presenza di difetti congeniti del sistema genito-urinario o di casi di tumore di Wilms in famiglia. Successivamente, viene eseguita una visita medica per verificare, tra le altre cose, la presenza di una massa palpabile nell'addome e per misurare la pressione sanguigna.
Per ottenere una conferma della diagnosi e valutarne l'estensione, il medico richiede degli esami di diagnostica per immagini. Il primo esame in genere consiste in un'ecografia dell'addome completo. Questa tecnica, non dolorosa e senza alcun fastidio per il bambino, permette di visualizzare i reni e altri organi addominali che potrebbero essere interessati dal tumore. L'ecografia dura circa 10-15 minuti: il bambino viene fatto sdraiare su un lettino e l'ecografista, dopo aver applicato sulla cute un gel che facilita l'esame, poggia la sonda sulla parte da studiare, facendola scorrere in diverse direzioni mentre osserva su uno schermo le immagini prodotte dagli ultrasuoni riflessi.
Altre analisi strumentali cruciali per la diagnosi sono la tomografia computerizzata (TC) e la risonanza magnetica (RM) dell'addome. Questi esami consentono di ipotizzare la natura del tumore, oltre che di valutarne l'eventuale crescita nelle strutture che circondano il rene o la diffusione in altri organi. La risonanza magnetica fornisce immagini molto precise delle parti interne, inclusi i tumori, utilizzando campi magnetici, senza esposizione a radiazioni ionizzanti. Tuttavia, l'esame richiede un tempo piuttosto lungo durante il quale il paziente deve rimanere immobile. La TC, invece, utilizza i raggi X per ottenere immagini dei tessuti interni e, a differenza di una semplice radiografia, permette di visualizzare anche i tessuti molli; espone però a radiazioni ionizzanti. Lo studio del torace attraverso la TC è particolarmente utile per valutare i polmoni, che sono la sede più frequente di comparsa di metastasi. Per l'esecuzione di questi esami, si possono somministrare ai bambini dei sedativi per mantenerli addormentati e quindi fermi per il tempo necessario. Questi esami vengono eseguiti in modo nativo e con la somministrazione di mezzo di contrasto. Possono essere integrati dall'urografia escretoria. I reperti caratteristici che di solito permettono di formulare la diagnosi sono mostrati in apposite panoramiche. La diffusione del tumore viene studiata con indagini strumentali (ecografie e TAC addominale, analizzando in particolare il fegato e il rene controlaterale), che sono anche utili per orientare il protocollo della chemioterapia post-operatoria.
Nella maggior parte dei casi, attraverso l'anamnesi, la visita medica e gli esami radiologici sopra indicati, è possibile formulare l'ipotesi diagnostica di tumore di Wilms con un basso margine di errore, rendendo non necessaria l'esecuzione di una biopsia. Solo in casi selezionati i medici potranno decidere di ricorrere alla biopsia con ago, al fine di analizzare un frammento di tumore al microscopio (analisi istopatologica). È importante sottolineare che, naturalmente, esiste un certo rischio di diagnosi errata quando ci si affida esclusivamente alla diagnostica per immagini. Se sussiste un dubbio intraoperatorio sulla diagnosi di nefroblastoma, si può prendere in considerazione una biopsia o una puntura del tumore solo se il tumore è considerato inoperabile; altrimenti, è obbligatorio procedere come se fosse uno stadio III.
Le analisi del sangue e delle urine danno informazioni sullo stato di salute dei reni, anche se solo eccezionalmente risultano alterate. Il dosaggio delle catecolamine urinarie, la cui concentrazione urinaria è normale nel nefroblastoma, può però aiutare a distinguere il tumore di Wilms dal neuroblastoma, un tumore che spesso si forma nelle ghiandole surrenali e che può presentarsi con caratteristiche simili al tumore di Wilms in bambini della stessa fascia di età, ma che raramente invade il rene per contiguità. La diagnosi differenziale si pone anche con altri tumori del rene, compreso il nefroma mesoblastico (soprattutto nella prima infanzia), il sarcoma a cellule chiare, i rabdomiomi e i tumori stromali metanefrici.
Caratteristiche Patologiche e Classificazione: Comprendere la Natura del Tumore
La diagnosi definitiva di nefroblastoma viene posta con la microscopia, che non solo consente di confermare la natura della neoplasia, ma permette anche di effettuare la stadiazione del tumore e di orientare la scelta della chemioterapia post-operatoria. Un patologo esamina sottili sezioni del tumore al microscopio per ricercare le tre componenti tipiche già menzionate: cellule blastemali (primitive), epiteliali (tubulari) e stromali (mesenchimali). Non tutti i tumori contengono tutte e tre le componenti. La classificazione istopatologica rimane il fondamento per la diagnosi finale di un tumore di Wilms. Macroscopicamente, il tumore di Wilms si presenta come una massa solida che può attraversare il rene.
Una caratteristica speciale e clinicamente rilevante è l'anaplasia, che si osserva in circa il 7-10% dei nefroblastomi. L'anaplasia indica che le cellule tumorali sono molto più grandi e anomale del normale, con figure di divisione cellulare distorte. L'anaplasia si divide in focale, quando è presente in aree limitate del tumore, e diffusa, quando è estesa in tutto il tumore. I tumori con anaplasia diffusa sono considerati più aggressivi e sono più difficili da curare rispetto a quelli in cui l'anaplasia è assente.
Per confermare la diagnosi di nefroblastoma, i patologi possono utilizzare un test chiamato immunoistochimica (IHC). Le cellule epiteliali, ad esempio, sono positive per citocheratina, EMA (antigene di membrana epiteliale) e CD56. La proteina WT1 è particolarmente importante in questo contesto, in quanto è presente in circa il 90% dei nefroblastomi.
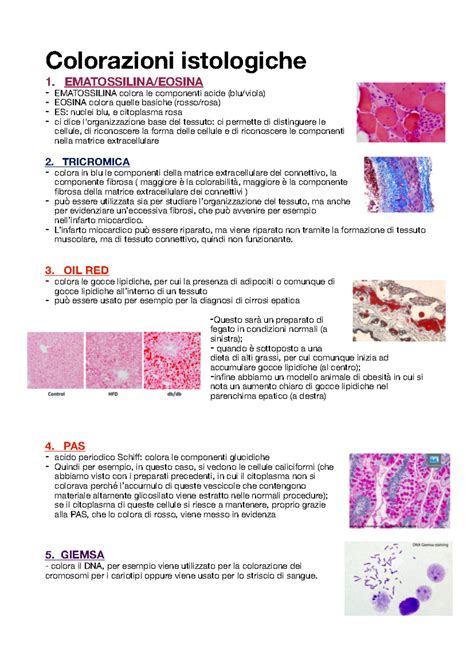
La classificazione del tumore consiste nel raggruppare i tumori in base al loro aspetto al microscopio. Questa classificazione è di fondamentale importanza per il nefroblastoma perché aiuta a prevedere il comportamento del tumore e a determinare la quantità e il tipo di trattamento necessario. Esistono due principali approcci di studio e classificazione del nefroblastoma a livello internazionale, che riflettono diverse strategie terapeutiche:
- Approccio della SIOP (Società Internazionale di Oncologia Pediatrica): Questo sistema classifica il tumore istologicamente dopo la somministrazione della chemioterapia pre-operatoria (neoadiuvante). Per i pazienti pretrattati con chemioterapia, la classificazione SIOP segue una versione della Classificazione di Stoccolma del 1995, rivista nel 2002.
- Approccio del NWTS (National Wilms Tumour Study Group): Questo sistema classifica il tumore prima della chemioterapia, poiché l'intervento chirurgico viene eseguito per primo. I tumori vengono classificati come a bassa, intermedia o alta malignità, oppure come a istologia favorevole (“favorable”) o sfavorevole (“unfavorable”).
Le differenze tra le due classificazioni si basano quindi sul diverso approccio terapeutico iniziale.
Stadiazione del Nefroblastoma: Valutare l'Estensione della Malattia
La stadiazione descrive quanto il tumore si è diffuso nell'organismo al momento della diagnosi ed è un fattore cruciale, in quanto guida la scelta del trattamento e aiuta a predire la prognosi. La stadiazione del tumore di Wilms viene formulata dopo l'intervento chirurgico di asportazione del rene con il tumore, ed è stabilita in base alla diffusione del tumore al di fuori dell'organo di origine (il rene). La stadiazione richiede una descrizione esatta dell'estensione intraoperatoria del tumore, che viene integrata dall'esame istologico. Secondo i criteri SIOP, ciò avviene dopo la chemioterapia preoperatoria. La determinazione concreta dello stadio tumorale è di notevole importanza clinica per la successiva stratificazione del rischio e la pianificazione della terapia. Lo stadio locale (addominale) del tumore primario deve essere sempre indicato.
Il sistema di stadiazione prevede cinque stadi principali:
- Stadio I: In questo stadio, il tumore è confinato esclusivamente al rene ed è stato completamente asportato chirurgicamente. Questo indica una malattia localizzata e completamente resecabile.
- Stadio II: Il tumore si estende oltre i confini del rene, ma è stato comunque completamente asportato. Ciò significa che, pur essendosi esteso localmente, non c'è più tumore residuo visibile dopo l'intervento.
- Stadio III: L'intervento chirurgico non è riuscito ad asportare tutto il tumore, lasciando del tumore residuo. Questo può essere dovuto al fatto che il tumore si è diffuso ai linfonodi addominali o si è rotto durante l'intervento chirurgico.
- Stadio IV: Questo stadio indica che il tumore si è diffuso in altri organi, manifestando metastasi a distanza. Le sedi più comuni di metastasi sono i polmoni, ma possono essere coinvolti anche il fegato o linfonodi distanti dal tumore primario.
- Stadio V: Questo è il caso di un coinvolgimento bilaterale dei reni, ovvero il tumore è presente in entrambi i reni al momento della diagnosi.

Strategie Terapeutiche: Un Approccio Multimodale e Personalizzato
La cura del tumore di Wilms prevede un approccio combinato che integra chirurgia, chemioterapia e, solo in alcuni casi selezionati, radioterapia. È fondamentale che interventi di questo tipo vengano eseguiti in centri altamente specializzati, con esperienza consolidata nella gestione di tumori pediatrici, per garantire la massima efficacia e sicurezza. Il trattamento dei bambini con tumore di Wilms ha senso solo se basato su protocolli di trattamento standardizzati e condivisi a livello internazionale.
Tumore di Wilms: guarire salvando la funzionalità renale
Chemioterapia
Il trattamento farmacologico rappresenta un pilastro fondamentale della terapia. Il nefroblastoma di solito risponde bene al trattamento con i citostatici, per questo la chemioterapia è una parte essenziale del regime terapeutico. Sempre più spesso, prima dell'intervento chirurgico, si esegue una fase di chemioterapia pre-operatoria, detta anche neoadiuvante. Questa strategia, oggi condivisa a livello internazionale e adottata principalmente secondo i protocolli SIOP, consente di ridurre le dimensioni del tumore, rendendo l'intervento più agevole e riducendo il rischio di complicanze intraoperatorie, incluse le rotture tumorali, il cui tasso è stato ridotto a meno del 5%. La chemioterapia preoperatoria consente di aumentare la percentuale di pazienti con uno stadio locale I post-intervento fino al 60%. Generalmente, viene somministrata a tutti i pazienti di età superiore ai sei mesi e inferiore ai 16 anni, purché la diagnosi per immagini sia certa. Nei pazienti con un tumore bilaterale, la chemioterapia preoperatoria è personalizzata, con l'obiettivo primario di poter eseguire una resezione del tumore che preservi il rene.
I farmaci più utilizzati sono la vincristina, l'actinomicina-D e la doxorubicina (nota anche come adriamicina), che si sono dimostrati molto efficaci nel ridurre e controllare la malattia. Nei casi più complessi, come quelli con istologia sfavorevole o con resistenza ai farmaci standard, possono essere utilizzati altri chemioterapici, come ifosfamide, etoposide e carboplatino, capaci di indurre una remissione anche in situazioni più difficili.
Il trattamento farmacologico prosegue anche dopo l'intervento chirurgico, e in questa fase è chiamato chemioterapia adiuvante. La chemioterapia viene effettuata a cicli: la somministrazione dei farmaci viene interrotta per poi essere ripresa a distanza di qualche tempo, in modo che l'organismo abbia modo di riprendersi. La durata e l'intensità dei cicli di chemioterapia dopo l'asportazione chirurgica dipendono dallo stadio del tumore e da alcune caratteristiche istologiche riscontrate all'esame al microscopio del pezzo asportato. Infatti, oltre all'eventuale presenza di anaplasia, i medici possono studiare se il tumore è regredito molto o poco per effetto della cura pre-operatoria, e ottenere indicazioni importanti sull'efficacia dei farmaci usati.Le strategie terapeutiche vengono adattate allo stadio della malattia. Nei bambini con stadio I, II o III e istologia favorevole, l'associazione di vincristina e actinomicina-D somministrata per un periodo variabile tra due e sei mesi rappresenta il trattamento di riferimento. Quando la malattia è più estesa, come nel caso degli stadi IV (con metastasi a distanza) o V (coinvolgimento bilaterale dei reni), oppure quando il tumore presenta caratteristiche istologiche più aggressive (alta malignità, istologia sfavorevole), il trattamento diventa più complesso e viene personalizzato in base al singolo caso, con terapie più intense e protocolli specifici.Nei pazienti con stadio iniziale IV, si distingue tra i "responders", in cui le metastasi non sono più rilevabili dopo sei settimane di chemioterapia preoperatoria, e i "non-responders", in cui le metastasi sono ancora presenti. I responsivi vengono trattati nel post-operatorio in base all'istologia e allo stadio, e la radioterapia delle metastasi polmonari non viene eseguita. Per i non-responders, la chemioterapia postoperatoria viene intensificata, ed è sempre necessaria l'esecuzione di una rimozione completa delle metastasi. L'importanza della chemioterapia ad alto dosaggio con trapianto autologo di cellule staminali (ABMT) nel trattamento dei pazienti ad alto rischio con tumore di Wilms non è ancora chiara e non può quindi essere raccomandata in generale.
Chirurgia
L'intervento chirurgico, che consiste nell'asportazione del rene colpito (nefrectomia) insieme ad alcuni linfonodi regionali, rappresenta uno degli atti terapeutici centrali nel trattamento di questo tumore. La nefrectomia del tumore ha due scopi principali: il tumore di Wilms deve essere asportato radicalmente, puntando a una resezione R0 (ovvero l'asportazione completa del tumore senza margini residui macroscopicamente rilevabili), ed è altrettanto importante determinare la diffusione del tumore, cioè lo stadio, in fase intraoperatoria. In genere, la rimozione del rene avviene in blocco, con grande attenzione alla radicalità dell'intervento e alla sicurezza del paziente. I principi della nefrectomia tumorale, stabiliti negli anni '50 da William Ladd e tuttora validi, prevedono un approccio transaddominale, un'esposizione accurata del rene colpito, evitando assolutamente la rottura del tumore, la resezione dei vasi renali e il posizionamento dell'uretere il più vicino possibile alla vescica. L'ispezione della cavità addominale prima della resezione del tumore è obbligatoria.

La dissezione linfonodale locoregionale, o "campionamento linfonodale", è indispensabile per determinare lo stadio del tumore e quindi è parte integrante della stratificazione del rischio, della terapia post-operatoria e della prognosi del paziente. Bisogna tenere presente che un tumore renale bilaterale viene diagnosticato con un alto grado di certezza se la diagnostica per immagini preoperatoria è di buona qualità. Pertanto, se il rene controlaterale è poco appariscente all'imaging, l'esposizione di questo rene può essere omessa durante l'intervento.
In situazioni particolari, come nel caso di bambini che presentano la malattia in entrambi i reni oppure che sono affetti da sindromi genetiche predisponenti, l'intervento può essere di tipo conservativo, noto come nefrectomia parziale. In questi casi, si cerca di preservare parte del rene colpito, per mantenere quanto più possibile la funzione renale. Tale procedura è eseguita in tutti i casi in cui sia anatomicamente possibile, compresi quelli di tumore monolaterale, e consente di salvaguardare quanto più parenchima renale sano. È molto importante che interventi chirurgici di nefrectomia parziale siano effettuati solo in centri che hanno una grossa esperienza nel gestire interventi chirurgici così complessi. Un individuo può condurre una vita normale anche con un rene solo, ma se vengono asportati entrambi i reni (una situazione che oggi si verifica solo eccezionalmente) è necessario ricorrere alla dialisi finché non è possibile effettuare un trapianto di rene. I nuovi concetti di terapia chirurgica includono l'aspetto della resezione del tumore che preserva il rene, giustificata dal rischio di nefroblastomi metacroni bilaterali, successiva ipertensione renale, perdita dell'organo controlaterale a causa di un trauma e compromissione della funzione renale da chemioterapia intensiva, ma che devono essere bilanciati con i criteri oncologici di radicalità.
Le trombosi vascolari, come estensioni del tumore nella vena renale e nella vena cava inferiore, sono ben note nel tumore di Wilms. Le estensioni che si protraggono sottodiaframmaticamente o oltre nell'atrio destro rappresentano una sfida chirurgica speciale. Questi dovrebbero essere operati dopo la chemioterapia preoperatoria, poiché la chirurgia primaria di tali trombi tumorali è stata associata a una maggiore morbilità. Il pretrattamento citostatico può ottenere una significativa riduzione delle dimensioni e dell'estensione del trombo tumorale, facilitando così la resezione. Un trombo che si estende a livello sopradiaframmatico o atriale deve essere operato sotto bypass extracorporeo con una macchina cuore-polmone, preferibilmente in condizioni di basso flusso o in arresto cardiaco, in ipotermia. Un altro aspetto della chirurgia del tumore di Wilms è la crescente importanza della resezione minimamente invasiva e laparoscopica del tumore.
Radioterapia
La radioterapia, invece, trova oggi un impiego più limitato rispetto al passato. Il tumore di Wilms è un tumore altamente radiosensibile. Con il progressivo sviluppo dei protocolli di trattamento SIOP e le conoscenze acquisite, in particolare lo sviluppo di combinazioni chemioterapiche efficaci, l'indicazione per la radioterapia potrebbe essere resa sempre più restrittiva. Viene riservata a casi selezionati, in particolare quando la malattia si presenta in fase avanzata o con localizzazioni metastatiche (come al polmone o al fegato), quando sono presenti dimensioni più ampie o in quelli che mostrano un'istologia meno favorevole. Ad esempio, nei pazienti con malattia allo stadio III e istologia favorevole, la combinazione tra chemioterapia e radioterapia mirata sull'area addominale può aumentare l'efficacia del trattamento. In questi casi, può essere aggiunta anche la doxorubicina per rafforzare l'effetto dei farmaci principali.
Prognosi e Follow-up: Alte Percentuali di Guarigione
Grazie all'adozione di protocolli terapeutici condivisi a livello internazionale e alla presa in carico da parte di centri specializzati, oggi le possibilità di guarigione per i bambini con tumore di Wilms sono molto alte, con percentuali di successo superiori al 90%. Il tumore di Wilms è infatti un ottimo esempio di malattia maligna quasi curabile, rappresentando una delle neoplasie pediatriche con la prognosi migliore. In Italia, circa il 92% dei bambini che si ammalano di tumore di Wilms è vivo a 5 anni dalla diagnosi. La sopravvivenza complessiva con tumore unilaterale senza metastasi è del 98%, mentre la sopravvivenza libera da recidiva è dell'88%.
La probabile evoluzione della malattia (prognosi) dipende soprattutto dallo stadio del tumore alla diagnosi e dall'eventuale presenza di anaplasia. I pazienti con malignità bassa e intermedia con tumori non metastatici hanno una prognosi di sopravvivenza superiore al 90%. Al contrario, i pazienti con anaplasia diffusa o con un sottotipo ricco di blastema presentano una prognosi significativamente meno favorevole. Se sono presenti metastasi a distanza, la prognosi dei pazienti dipende in modo decisivo dalla risposta alla chemioterapia effettuata. Inoltre, diversi marcatori genetici (come la LOH di 11q, 16q, 22q e le mutazioni di p53) sembrano essere correlati a una prognosi sfavorevole.
È possibile che la malattia si ripresenti (recidiva). In questi casi, la prognosi è più favorevole se alla diagnosi il tumore era in stadio I-II, l'anaplasia era assente e se per la chemioterapia erano stati usati solo alcuni farmaci. Un'ulteriore intensificazione del trattamento non porta a un ulteriore miglioramento per la maggior parte dei pazienti, ma questo contrasta con una minoranza di pazienti che hanno una prognosi significativamente peggiore. Gli studi di ottimizzazione della terapia mirano all'ulteriore miglioramento della terapia, alla riduzione degli effetti collaterali e degli effetti tardivi della malattia, a una migliore stratificazione del rischio e, in ultima analisi, all'ulteriore miglioramento della prognosi del nefroblastoma.
Prevenzione e Screening: Individuare Precocemente i Rischi
Attualmente, non esistono strategie preventive specifiche ed efficaci per il tumore di Wilms. Ogni attività di prevenzione primaria richiede che le cause del tipo di tumore che si desidera prevenire siano note, a livello di popolazione, almeno con i dati statistici delle osservazioni epidemiologiche, e possibilmente anche con i possibili meccanismi biologici messi in luce tramite esperimenti di laboratorio. Tuttavia è raro che i tumori abbiano una singola causa, e per questo nella stragrande maggioranza dei casi è difficile se non impossibile stabilire a posteriori, con criteri scientifici, l'origine di un tumore che è insorto in un individuo. È difficilissimo negli adulti, per i quali a volte si può soltanto presumere che l'esposizione a sostanze cancerogene o abitudini e comportamenti non salutari possano avere contribuito alla crescita tumorale. È ancora più difficile nei bambini, data la giovane età.
Per i tumori di Wilms non è al momento possibile definire strategie efficaci per la prevenzione, dal momento che l'epidemiologia non ha a oggi identificato fattori di rischio modificabili. Per questo è importante sottolineare che, qualora un tumore di Wilms insorga in un bambino, i genitori non si devono rimproverare nulla per la malattia del proprio figlio.
Tuttavia, nei bambini con sindromi genetiche note per aumentare il rischio di questa neoplasia, è fortemente consigliato un monitoraggio regolare. Questo avviene attraverso ecografie addominali periodiche, al fine di individuare precocemente eventuali anomalie. I bambini che presentano sindromi genetiche associate al rischio di sviluppare un tumore di Wilms vengono sottoposti a visite di controllo e a ecografie su base regolare, in modo da identificare il tumore il prima possibile, seguendo le raccomandazioni dei medici genetisti.