La genetica umana rappresenta una rete complessa di istruzioni biologiche in cui anche minime variazioni possono portare a conseguenze cliniche significative. Quando parliamo di condizioni che coinvolgono l'integrità cromosomica, come la sindrome di Edwards, o di patologie che colpiscono i sensi, come la sindrome di Usher e le forme di sordità genetica, ci addentriamo in un panorama in cui la diagnostica molecolare e la ricerca clinica lavorano per migliorare la prognosi e la qualità della vita dei pazienti.
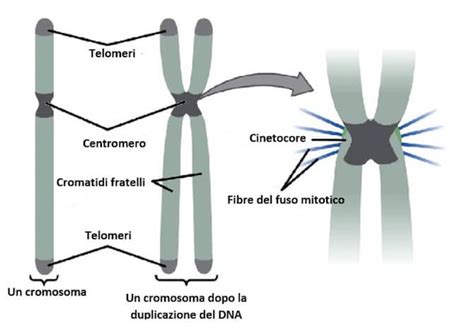
Le diverse forme di Trisomia 18 (Sindrome di Edwards)
La sindrome di Edwards, nota anche come trisomia 18, è una condizione cromosomica causata dalla presenza di una copia extra del cromosoma 18. Questa anomalia si manifesta con gradi di gravità differenti a seconda della modalità con cui il materiale genetico in eccesso è distribuito nell'organismo.
La forma completa è la tipologia più frequente, interessando circa il 94% dei bambini affetti da sindrome di Edwards. In questo quadro, ogni cellula del corpo possiede tre copie del cromosoma 18, invece delle consuete due. Questa presenza ubiquitaria del materiale genetico extra determina le caratteristiche cliniche tipiche della sindrome.
La trisomia a mosaico, che colpisce circa il 5% dei bambini, rappresenta una variante in cui la copia extra del cromosoma 18 è presente solo in alcune cellule del corpo. La gravità dei segni e dei sintomi della malattia a mosaico dipende strettamente dal tipo e dal numero di cellule che hanno il cromosoma supplementare. Di conseguenza, alcuni bambini possono essere colpiti solo lievemente dalla malattia, mentre altri lo sono in modo molto grave.
La forma parziale è la meno frequente tra i tipi di trisomia 18. In questa condizione, ad essere triplicato non è l'intero cromosoma, ma solo un segmento specifico. La variabilità clinica tra queste forme evidenzia come l'impatto di un'aneuploidia non sia solo quantitativo, ma dipenda profondamente dalla distribuzione cellulare dell'anomalia.
Diagnostica prenatale e screening delle aneuploidie
La moderna medicina prenatale offre numerosi strumenti per identificare le anomalie cromosomiche precocemente. Il percorso diagnostico inizia solitamente con il test combinato, eseguito tra l'11a e la 14a settimana di gestazione. Questo screening comprende un esame specifico di sangue e il test della translucenza nucale, un'ecografia mirata per la ricerca dell'ispessimento cutaneo nella parte posteriore del collo del bambino.
Per giungere a una diagnosi di certezza, si ricorre a indagini invasive. L'amniocentesi rimane l'indagine principale per lo studio dei cromosomi fetali e la diagnosi prenatale delle aneuploidie cromosomiche. Eseguita intorno alla 16a-18a settimana, consente il prelievo del liquido amniotico dalla cavità uterina attraverso la parete addominale.
Alternativamente, la villocentesi, o prelievo dei villi coriali, è una tecnica per la diagnosi prenatale più invasiva dell'amniocentesi. Si esegue più precocemente, intorno alla 10a-11a settimana, e consiste nel prelievo per via transaddominale e sotto controllo ecografico di tessuto trofoblastico, contenente cellule fetali. Sul campione si può eseguire un'analisi cromosomica diretta o, come nel caso del liquido amniotico, le cellule sono poste in coltura e successivamente analizzate.
Di recente, è stato messo a punto un test che prevede l'analisi del DNA fetale, noto come screening prenatale non invasivo (NIPT). Ottenuto da un campione di sangue materno dalla 10a settimana in avanti, permette di effettuare esami citogenetici e molecolari riducendo i rischi legati alle procedure invasive.
Un'introduzione alla genetica e al test genetico prenatale
La complessità della sindrome di Usher
La sindrome di Usher è una rara malattia genetica che colpisce sia l'udito che la vista, causando notevoli difficoltà nella vita quotidiana. È la condizione più comune che causa sordità e cecità combinate, con un impatto profondo sugli individui e sulle loro famiglie. Comprendere la sindrome di Usher è fondamentale per una diagnosi precoce, una gestione efficace e il miglioramento della qualità della vita delle persone colpite.
Questa patologia è caratterizzata dalla combinazione di perdita dell'udito e progressiva perdita della vista dovuta a retinite pigmentosa (RP). È classificata in tre tipi principali (Tipo 1, Tipo 2 e Tipo 3), ciascuno con diversi gradi di gravità ed età di insorgenza. La malattia è causata da mutazioni in geni specifici, come MYO7A, USH2A e CDH23, ereditati con modalità autosomica recessiva. Ciò significa che un individuo deve ereditare due copie del gene mutato, una da ciascun genitore, per sviluppare la sindrome.
Il Tipo 1 si presenta tipicamente con una grave perdita dell'udito alla nascita e una progressiva perdita della vista durante l'infanzia. Sebbene non esistano cure definitive, la ricerca scientifica sta esplorando nuove vie. In Germania, i ricercatori della Johannes Gutenberg University di Mainz, guidati dal professor Uwe Wolfrum, hanno individuato due possibili strategie terapeutiche per la mutazione del gene USH1C: la riparazione del DNA tramite nucleasi "a forbice molecolare" (Zn-finger) e l'utilizzo di molecole in grado di "saltare" i segnali di stop prematuri nel gene, come il PTC124 (Ataluren).
Sordità genetica sindromica e non sindromica
La sordità genetica sindromica identifica un gruppo eterogeneo di disturbi dell'udito di origine ereditaria che si manifestano in associazione ad altre anomalie cliniche o malformazioni. Queste condizioni rappresentano circa il 30% di tutti i casi di sordità genetica prelinguale. Esistono centinaia di sindromi associate alla sordità, tra cui quelle che coinvolgono anomalie della pigmentazione, difetti renali, o problemi cardiaci.
La trasmissione può avvenire in modalità autosomica recessiva (la forma più comune), autosomica dominante o legata al cromosoma X. I fattori di rischio principali sono legati alla storia familiare, e la consanguineità tra i genitori aumenta significativamente il rischio di manifestare forme recessive.
Sul fronte della sordità non sindromica, la classificazione internazionale utilizza le sigle DFNA (autosomica dominante), DFNB (autosomica recessiva) e DFN (X-linked). Un ruolo cruciale è svolto dalle connessine, proteine di membrana che formano complessi definiti connessoni. Ad esempio, nel gene della connessina 26 sono state descritte oltre 100 varianti differenti; nelle popolazioni caucasiche, la variante più frequente è la 35delG. Un'altra proteina fondamentale è l'otoferlina, che interviene nella regolazione delle vescicole presinaptiche delle cellule cigliate interne.
Il percorso diagnostico deve essere tempestivo e multidisciplinare. I test genetici, attraverso il sequenziamento di nuova generazione (NGS), permettono di analizzare contemporaneamente centinaia di geni noti per essere associati alla sordità. Per quanto riguarda la gestione, l'impianto cocleare rappresenta la soluzione d'elezione per i bambini con ipoacusia neurosensoriale profonda, permettendo spesso, se abbinato a un percorso logopedico intensivo, di raggiungere livelli di linguaggio verbale paragonabili ai coetanei udenti.
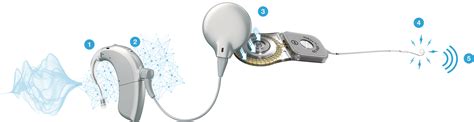
La ricerca continua a progredire, con studi recenti come quelli pubblicati su Ann Hum Genet e Ear Hear che approfondiscono il modello di previsione degli esiti audiologici nei pazienti portatori di mutazioni specifiche, confermando che la comprensione molecolare del difetto è la chiave per una personalizzazione sempre più accurata del trattamento terapeutico.