La sindrome di Down, nota scientificamente come trisomia 21, è una condizione genetica complessa e la più comune anomalia cromosomica che causa disabilità intellettiva. Essa è caratterizzata dalla presenza di una copia extra, totale o parziale, del cromosoma 21. Questa anomalia cromosomica, dimostrata per la prima volta nel 1959, altera il dosaggio genico e modifica l’espressione di centinaia di geni, influenzando molteplici reti cellulari durante lo sviluppo e nell'età adulta. Le conseguenze di questa alterazione si manifestano con caratteristiche fisiche peculiari e modifiche nello sviluppo, la cui gravità può variare notevolmente da individuo a individuo. Sebbene la maggior parte dei casi di sindrome di Down sia attribuita a un errore nella divisione cellulare noto come non-disgiunzione, una percentuale significativa, pur se minore, è dovuta a una specifica anomalia strutturale: la traslocazione robertsoniana. Comprendere le cause sottostanti, le diverse forme della sindrome, le sue manifestazioni cliniche e gli avanzamenti nella diagnosi e nella gestione è fondamentale per migliorare la qualità di vita delle persone affette e promuovere la loro piena inclusione sociale.
Fondamenti Genetici: I Cromosomi e la Trisomia 21
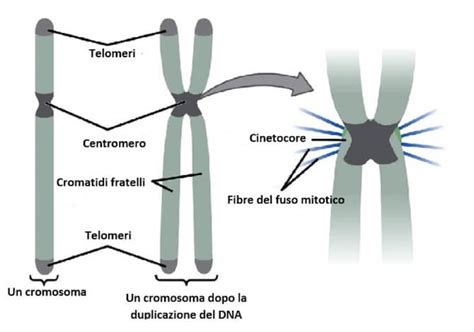
Per comprendere la sindrome di Down, è essenziale iniziare dai concetti fondamentali della genetica. All'interno delle cellule umane, strutture dette cromosomi contengono i geni, che sono segmenti di acido deossiribonucleico (DNA) contenenti il codice di una specifica proteina. Ogni cellula umana, ad eccezione di spermatozoi e ovuli, contiene normalmente 46 cromosomi, organizzati in 23 coppie. Di queste, 22 coppie sono autosomiche, ovvero non correlate al sesso, mentre una coppia è costituita dai cromosomi sessuali (XX per le femmine e XY per i maschi). I gameti, cioè le cellule riproduttive come spermatozoi e ovuli, destinati ad unirsi, contengono invece 23 cromosomi, ovvero la metà. Un cromosoma di ogni coppia è ereditato dalla madre e l'altro dal padre.
Le anomalie cromosomiche si distinguono in due categorie principali: numeriche e strutturali. Le anomalie numeriche si verificano quando un individuo possiede una o più copie extra di un cromosoma - in questo caso si parla di trisomia - o un cromosoma mancante, definito monosomia. Le anomalie strutturali, invece, si presentano quando una parte di un cromosoma risulta anomala. Talvolta, un cromosoma intero o una sua porzione si unisce in modo errato con un altro, fenomeno noto come traslocazione. In altri casi, parti di cromosomi possono risultare mancanti (delezione) o duplicate. Alcune di queste anomalie possono causare la morte dell’embrione o del feto prima della nascita.
La più comune trisomia riscontrabile in un neonato è la trisomia 21, che è la causa della stragrande maggioranza, circa il 95%, dei casi di sindrome di Down. Questa condizione deriva dalla presenza di tre copie complete del cromosoma 21, anziché le usuali due, in tutte le cellule dell’organismo. Tale eccesso comporta la sovra-espressione di numerosi geni localizzati sul braccio lungo del cromosoma 21 (21q), tra cui, in letteratura, DYRK1A, APP, RCAN1, ETS2, SOD1, i recettori dell’interferone e alcuni microRNA. L'effetto di questo sovradosaggio genico non è limitato ai soli geni "in più"; piuttosto, l'omeostasi cellulare viene ricalibrata, con riorganizzazioni trascrittomiche ed epigenetiche che interessano anche altri cromosomi. Queste alterazioni colpiscono processi chiave come la neurogenesi e la maturazione sinaptica, il metabolismo ossidativo e la funzione mitocondriale, la risposta immunitaria e infiammatoria, la morfogenesi cardiaca e l'emopoiesi.
Le Cause della Sindrome di Down: Oltre la Non-Disgiunzione
La sindrome di Down può presentarsi in diverse forme citogenetiche, ciascuna con specifiche implicazioni in termini di cause e rischi di ricorrenza. Le tre forme principali sono la trisomia 21 libera, il mosaicismo e la traslocazione cromosomica.
La Trisomia 21 Libera (Non-Disgiunzione)
La forma più frequente di sindrome di Down, responsabile di circa il 95% dei casi, è la trisomia 21 libera. Questa condizione si verifica a causa di un evento di non-disgiunzione, un errore nella divisione cellulare che avviene durante la formazione dei gameti (ovocita o spermatozoo). Normalmente, durante la meiosi, i due cromosomi di una coppia, provenienti ciascuno da uno dei genitori, si separano correttamente. Nel caso della non-disgiunzione, questa separazione non avviene come dovrebbe, il che può portare alla formazione di un gamete con una copia extra del cromosoma 21 o con un cromosoma 21 mancante. Se un gamete con un cromosoma 21 supplementare partecipa alla fecondazione, l'embrione risultante avrà tre copie del cromosoma 21 in ogni sua cellula.
Nella grande maggioranza dei casi di trisomia 21 libera, l'errore è di origine materna. Le cause precise responsabili della mancata disgiunzione cromosomica non sono ancora completamente chiarite, ma l'unico fattore di rischio importante e ampiamente riconosciuto è l'età materna avanzata. Il rischio di una coppia di avere un bambino con un cromosoma supplementare aumenta gradualmente con l'età della madre, divenendo considerevole dopo i 35 anni. Sebbene la prevalenza delle anomalie cromosomiche sia più alta nei feti nelle prime settimane di gestazione rispetto ai bambini nati vivi, a causa dell'elevata incidenza di aborti spontanei, la sindrome di Down può verificarsi in gravidanze di donne di ogni età. Tuttavia, statisticamente, la maggior parte dei bambini con sindrome di Down nasce da madri di età inferiore ai 35 anni, semplicemente perché le nascite in questa fascia d'età sono numericamente più frequenti. La non-disgiunzione è un evento che si verifica sporadicamente, rendendolo non prevedibile. Una madre che ha avuto un bambino affetto da trisomia 21 libera può dare alla luce un bambino perfettamente sano in una successiva gravidanza, sebbene un rischio lievemente aumentato persista (circa l'1% oltre al rischio legato all'età materna). L'età paterna ha un impatto minore sul rischio di non-disgiunzione.
Il Mosaicismo
Una variante assai più rara della sindrome di Down è il mosaicismo, che si presenta in circa l'1-2% dei casi. In questa forma, l'errore nella divisione cellulare avviene dopo il concepimento, durante le prime mitosi embrionali. Di conseguenza, solo alcune cellule dell'organismo presentano la trisomia 21 (ovvero tre cromosomi 21), mentre altre possiedono il corretto numero di due cromosomi 21. Le persone con sindrome di Down a mosaico hanno due linee cellulari: una con i normali 46 cromosomi e una con 47 cromosomi, inclusa una copia extra del cromosoma 21.
La prognosi per l'intelligenza e il rischio di complicanze mediche nelle persone con mosaicismo probabilmente dipendono dalla proporzione di cellule anormali (trisomia 21) in ciascun tessuto diverso, compreso il cervello. Tuttavia, in pratica, questo rischio non può essere previsto con certezza, poiché non è possibile determinare il cariotipo in ogni singola cellula del corpo. Alcune persone con mosaicismo della sindrome di Down possono avere segni clinici molto sottili e un'intelligenza considerata normale, ma anche chi non è affetto da mosaicismo può presentare una variabilità significativa nei reperti clinici.
La Traslocazione Robertsoniana: Una Causa Specifica e Ereditabile
La traslocazione cromosomica è responsabile di circa il 4% dei casi di sindrome di Down, e tra queste, la traslocazione robertsoniana è la più rilevante per la trisomia 21. A differenza della trisomia 21 libera, la traslocazione robertsoniana può essere ereditata da uno dei genitori, rappresentando l'unico caso in cui si può parlare di un errore ereditario nella sindrome di Down.
Una traslocazione è un'alterazione strutturale dei cromosomi che deriva da uno scambio di porzioni tra cromosomi non omologhi. Una traslocazione è definita bilanciata se lo scambio non comporta né la perdita né il guadagno di materiale genetico significativo. Le persone con un riarrangiamento cromosomico bilanciato sono, di solito, fenotipicamente normali, ovvero non manifestano i sintomi della sindrome, ma sono portatrici di un riarrangiamento.
La traslocazione robertsoniana consiste specificamente nella fusione di due cromosomi acrocentrici. I cromosomi acrocentrici nell’uomo sono il 13, 14, 15, 21 e 22, caratterizzati dal centromero situato molto vicino alla parte terminale. In una traslocazione robertsoniana, i due cromosomi si rompono entrambi in corrispondenza del centromero, ma su lati opposti. Per unione dei frammenti rotti si forma un nuovo cromosoma costituito dalle braccia lunghe dei due elementi originali, mentre le piccole braccia corte, geneticamente inerti, si uniscono a formare un piccolo cromosoma che di solito viene perso. In seguito a questa anomalia, il corredo cromosomico dell’individuo viene ridotto di un'unità, ma, poiché il materiale genetico perso è considerato inerte, il portatore non presenta conseguenze fenotipiche ed è quindi fenotipicamente normale. La traslocazione robertsoniana più comune coinvolge i cromosomi 13 e 14, rilevata con una frequenza di circa 1 su 1300 nati.
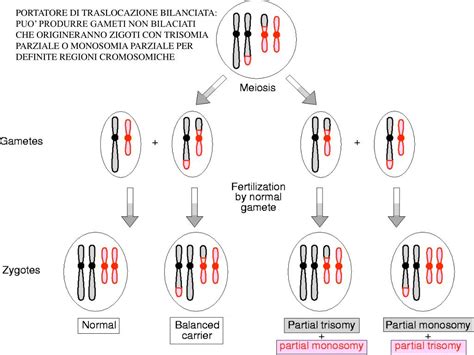
Nel contesto della sindrome di Down, la traslocazione robertsoniana più comune è la t(14;21), in cui il cromosoma 21 è attaccato al cromosoma 14. Questa è una traslocazione sbilanciata che, se presente nella prole, comporta un cariotipo di 45 cromosomi con l'aggiunta di materiale del cromosoma 21, risultando nella trisomia 21.
- Origine e Rischi di Ricorrenza: Circa la metà dei soggetti con t(14;21) presenta una traslocazione insorta de novo, ovvero non presente in nessuno dei genitori, i quali hanno un cariotipo normale. Nell'altra metà dei casi, uno dei genitori (quasi sempre la madre) è un portatore bilanciato della traslocazione, pur non essendo affetto dalla sindrome di Down. Durante la gametogenesi, un genitore portatore bilanciato può formare gameti con materiale 21 in eccesso, aumentando il rischio di concepire un feto affetto.
- Se la madre è portatrice di una traslocazione t(14;21), il rischio reale di avere un bambino con sindrome di Down è di circa 1:10 (teoricamente potrebbe essere 1:3).
- Se il padre è il portatore di una traslocazione t(14;21), il rischio è inferiore, solo di 1:20.
- Un'altra traslocazione frequente è la t(21;22). In questi casi, le madri portatrici hanno un rischio di circa 1:10 di avere un bambino con sindrome di Down, mentre per i padri portatori il rischio è ancora minore.
- La traslocazione 21q21q, in cui un cromosoma 21 è collegato a un altro cromosoma 21, è molto meno comune ma comporta un rischio elevatissimo: un genitore portatore bilanciato di questa traslocazione ha una probabilità del 100% di avere un figlio affetto dalla sindrome di Down, poiché tutta la prole vitale risulterebbe con la sindrome di Down o con monosomia 21, quest'ultima tipicamente incompatibile con la vita.
È essenziale determinare se un genitore è portatore o mosaico per la traslocazione 21q21q per comprendere i rischi per i loro figli. Se la trisomia 21 è dovuta a traslocazione robertsoniana, è fortemente indicato lo studio del cariotipo dei genitori. Con genitori con cariotipo normale, il rischio residuo è in genere di circa il 2-3%. Le persone con anomalie cromosomiche nei genitori, inclusi i riarrangiamenti bilanciati, devono essere indirizzate a una consulenza genetica e si deve prendere in considerazione lo studio prenatale. Questo sottolinea l'importanza di una diagnosi genetica precisa per la pianificazione familiare e la consulenza sui rischi di ricorrenza.
Aneuploidie ed anomalie cromosomiche
Manifestazioni Cliniche e Caratteristiche della Sindrome di Down
La sindrome di Down si manifesta con un insieme di caratteristiche fisiche e alterazioni dello sviluppo che possono variare in gravità da individuo a individuo. Non tutte le manifestazioni sono presenti in ogni persona affetta, e la loro espressione può essere di grado lieve, moderato o severo, a seconda del danno genetico e della risposta individuale.
Caratteristiche Fisiche Distintive
I bambini e gli adulti con sindrome di Down presentano peculiari caratteristiche fisiche, sebbene risentano anche degli attributi individuali. Alla nascita, i neonati affetti tendono a essere tranquilli, piangono raramente e presentano ipotonia (scarso tono muscolare). La maggior parte ha un profilo facciale piatto, soprattutto a livello del ponte nasale, e l'occipite appiattito (microcefalia). Sono anche frequenti l'eccesso di cute nella parte posteriore del collo e una statura generalmente ridotta rispetto ai coetanei, con uno sviluppo in altezza che rimane inferiore.
Le caratteristiche fisiche tipiche includono:
- Occhi: Spesso mostrano un'angolazione diretta verso l'alto sul bordo laterale. Sono di solito presenti pieghe epicantali agli angoli interni degli occhi. Possono essere visibili le macchie di Brushfield, ovvero piccole macchie grigie o bianche simili a grani di sale, situate perifericamente rispetto all'iride.
- Bocca e Orecchie: La bocca è spesso mantenuta aperta, e una lingua protrusa e rugosa può non avere una scissura centrale. Le orecchie sono spesso piccole e arrotondate.
- Mani: Tendono a essere corte e larghe, e spesso presentano una sola piega palmare trasversale. Le dita sono spesso corte, con clinodattilia (incurvamento) del 5° dito, che spesso ha solo due falangi.
- Piedi: Possono presentare un'ampia distanza fra il primo e il secondo dito, un tratto noto come "dita a sandalo", e il solco plantare si prolunga spesso posteriormente sul piede.

Sviluppo Cognitivo e Neurologico
Con la crescita, il ritardo fisico e lo sviluppo mentale diventano evidenti nei bambini con sindrome di Down. La disabilità intellettiva varia ampiamente da lieve a moderata, ma raramente può essere grave, con un quoziente intellettivo (QI) che raggiunge in media un valore pari a 50, rispetto al 100 mediamente registrato nella popolazione generale. I bambini portatori di trisomia 21 possono manifestare ritardi nell’apprendimento delle abilità principali, quali il linguaggio, la capacità mnemonica a breve e lungo termine e le abilità motorie.
Nel corso dell’infanzia, si riscontra spesso un comportamento che indica disturbo da deficit di attenzione e iperattività (ADHD). L'incidenza di comportamenti associati al disturbo dello spettro autistico risulta aumentata, in particolare nei bambini con grave disabilità intellettiva. Vi è anche un aumentato rischio di depressione sia nei bambini che negli adulti con sindrome di Down.
Il processo di invecchiamento appare accelerato in queste persone. Sintomi simili alla malattia di Alzheimer, quali demenza, amnesia, peggioramento delle capacità intellettive e alterazioni della personalità, possono svilupparsi in età relativamente giovane.Una patologia rara ma letale che può coinvolgere le arterie carotidi in alcuni individui con sindrome di Down è la malattia di Moyamoya. In questa condizione, le arterie carotidi si restringono, riducendo l’afflusso di sangue al cervello. Il corpo tenta di compensare sviluppando una rete di piccoli vasi sanguigni collaterali alla base del cranio, che appaiono ai raggi X simili a "nuvole di fumo" ("moyamoya" in giapponese). Il sintomo principale di questa malattia è l'ictus, ma possono presentarsi anche cefalea, crisi epilettiche, depressione e disturbi cognitivi.
Comorbilità Mediche Frequenti
Le persone con sindrome di Down hanno un rischio più elevato di incorrere in diverse condizioni patologiche che possono influenzare la funzionalità di quasi tutti gli organi o sistemi fisiologici dell'organismo. Per questo motivo, è fondamentale un monitoraggio costante e un approccio multidisciplinare alla cura.
- Cardiopatie Congenite: Circa il 50% dei bambini con sindrome di Down nasce con difetti cardiaci congeniti. I difetti del setto interventricolare e i difetti del setto atrioventricolare (anche chiamati difetti dei cuscinetti endocardici o difetti del canale atrioventricolare) sono i più frequenti. Questi difetti possono variare in gravità e spesso richiedono controlli periodici e interventi chirurgici, anche nella prima infanzia. I neonati possono essere asintomatici o mostrare segni di insufficienza cardiaca come respiro affannoso, aumento della frequenza respiratoria, difficoltà di alimentazione, sudorazione e scarso aumento di peso.
- Anomalie Gastrointestinali: Circa il 6% delle persone affette presenta anomalie gastrointestinali. Tra queste, l'atresia duodenale (a volte con pancreas anulare) è notevole e può manifestarsi con vomito biliare. Altre condizioni più comuni includono la malattia di Hirschsprung (mancanza di cellule nervose che controllano parti del colon, causando ritardo nell'evacuazione del meconio e sintomi di occlusione intestinale), la celiachia, la stenosi ipertrofica del piloro e l'atresia anale.
- Endocrinopatie e Metabolismo: Molte persone sviluppano endocrinopatie, con malattie della tiroide (il più delle volte ipotiroidismo) e diabete più comuni. Vi è anche una maggiore tendenza a sviluppare obesità rispetto alla popolazione generale.
- Problemi Scheletrici: Le articolazioni del collo possono essere instabili a causa dell'ipermobilità atlanto-occipitale e atlanto-assiale, così come anomalie ossee della colonna cervicale. Questa instabilità può determinare una compressione del midollo spinale, portando a cambiamenti nell'andatura, nell'uso delle braccia e delle mani, nella funzione intestinale e vescicale o debolezza.
- Problemi Sensoriali:
- Vista: Circa il 60% delle persone ha problemi agli occhi, inclusa la cataratta congenita (opacità del cristallino presente alla nascita o poco dopo), glaucoma, strabismo ed errori di rifrazione. È fondamentale un esame completo della vista alla nascita.
- Udito: La maggior parte delle persone affette ha una perdita dell'udito. Le infezioni dell'orecchio (otiti ricorrenti) sono molto frequenti, spesso associate a disfunzione della tuba di Eustachio a causa di alterazioni nella base cranica. Lo screening di routine per i problemi dell'udito è altamente consigliato, e l'uso di apparecchi acustici può essere di grande aiuto nei casi più gravi.
- Rischio Oncologico: La trisomia 21 è associata a particolari diatesi leucemiche. La leucemia linfoblastica acuta è almeno 20 volte più frequente nei bambini con sindrome di Down, e la forma megacarioblastica lo è di almeno 500 volte.
- Fertilità: Le donne con sindrome di Down possono avere il 50% delle possibilità di avere un bambino con sindrome di Down, ma molte gravidanze con feti affetti si interrompono spontaneamente. Gli uomini affetti da sindrome di Down sono di solito sterili, salvo rari casi di sindrome di Down a mosaico.
L’aspettativa di vita delle persone con sindrome di Down è aumentata notevolmente negli ultimi decenni. Se negli anni Sessanta era di appena 15 anni, oggi, nelle nazioni ad alto reddito, supera mediamente i 55-60 anni, con alcuni pazienti che hanno superato i 70 e anche gli 80 anni. Questo miglioramento è dovuto principalmente ai progressi nella medicina e nell'assistenza sanitaria, che consentono di trattare efficacemente le complicanze.
Screening e Diagnosi della Sindrome di Down
La diagnosi della sindrome di Down ha compiuto passi da gigante, rendendo molto più facile e sicuro di un tempo scoprire con tempestività se un feto è portatore della sindrome. La diagnosi può avvenire in fase prenatale o postnatale, e si avvale di test di screening e test diagnostici.
Importanza della Diagnosi Prenatale e Consulenza Genetica
Lo screening e la diagnosi prenatale devono essere offerti a tutte le donne in gravidanza, indipendentemente dall'età. L'età materna avanzata è l'indicazione più frequente allo studio citogenetico prenatale, dato che il rischio di anomalie cromosomiche aumenta esponenzialmente dopo i 35 anni. Anche anomalie cromosomiche in un precedente figlio o la conoscenza di un genitore portatore di una traslocazione bilanciata sono indicazioni alla diagnosi prenatale.
Il consenso informato e la decisione condivisa sono pilastri fondamentali in questo percorso, così come la tutela dei dati genetici. Le persone con un riarrangiamento cromosomico bilanciato, come nel caso della traslocazione robertsoniana, dovrebbero essere inviate a una consulenza genetica per comprendere appieno i rischi per la prole e le opzioni disponibili.
Test di Screening Non Invasivi
I test di screening sono esami che rilevano un rischio o una probabilità che il nascituro sia portatore della sindrome di Down o di altra anomalia genetica, ma non forniscono una certezza diagnostica. Vengono eseguiti su tutte le donne incinte e vanno integrati con altri più specifici.
- Test del Sangue Materno (Biochimici): Rilevano la concentrazione di specifiche proteine e ormoni associati alla gravidanza.
- Nel primo trimestre (tra 11+0 e 13+6 settimane): PAPP-A (proteina plasmatica A associata alla gravidanza) e beta-hCG libera (gonadotropina corionica umana).
- Nel secondo trimestre (tra la 15ª e la 18ª settimana): Tri Test, che valuta alfa-fetoproteina (AFP), estriolo non coniugato (uE3) e frazione beta della gonadotropina corionica (hCG). Nei casi di sindrome di Down, i livelli di AFP e uE3 tendono a essere inferiori ai valori medi, mentre i livelli di hCG tendono a essere più alti.
- Ecografia Fetale:
- Translucenza Nucale (NT): Tra l'11ª e la 14ª settimana di gravidanza, si misura con gli ultrasuoni lo spessore di una specifica area della testa del feto dietro il collo. Un aumento di questo valore indica un rischio più elevato di sindrome di Down e altre anomalie fetali.
- Screening Combinato: L'integrazione del test della translucenza nucale con i marcatori biochimici materni (PAPP-A e hCG libera) offre una buona performance di screening per la trisomia 21 e altre aneuploidie, con valori di rilevazione attorno all'82-90% e falsi positivi del 3-5%. Vengono valutati anche altri marker ecografici come l'osso nasale, il flusso nel dotto venoso e il rigurgito tricuspide.
- Ecografia Morfologica: Tra 18 e 22 settimane, è raccomandata per tutte le gravidanze per la ricerca di malformazioni maggiori.
- Test del DNA Fetale (NIPT/cfDNA): Questo esame di recente introduzione, disponibile dalla 10ª settimana, si effettua tramite prelievo ematico materno. Analizza il DNA fetale privo di cellule (cfDNA) che circola nel sangue della madre. Per la trisomia 21, la sensibilità e la specificità sono molto elevate (generalmente >99%). Tuttavia, il cfDNA rimane uno screening: un risultato "ad alto rischio" deve sempre essere confermato con un test diagnostico invasivo prima di prendere decisioni cliniche importanti. Fattori come un BMI elevato o un prelievo troppo precoce possono ridurre la "fetal fraction" e aumentare il tasso di risultati indeterminati.
Test Diagnostici Invasivi (Conferma)
Se la sindrome di Down è sospettata in base ai test di screening, è raccomandato un test di conferma fetale o postnatale. Questi esami forniscono una diagnosi certa.
- Villocentesi (Prelievo dei Villi Coriali): Prevede il prelievo di cellule della placenta (villi coriali) per analizzare il DNA fetale. Si effettua in genere tra la 10ª e la 13ª settimana di gravidanza.
- Amniocentesi: Si effettua prelevando una minima quantità di liquido amniotico dalla gestante attraverso un ago inserito nell’utero, solitamente dal secondo trimestre (≥15 settimane). Il campione viene usato per analizzare i cromosomi delle cellule fetali.
- Cordocentesi (Prelievo Percutaneo di Sangue Ombelicale): È una procedura invasiva utilizzata per ottenere sangue fetale direttamente dal cordone ombelicale, consentendo un cariotipo rapido. È generalmente riservata a situazioni in cui altri metodi diagnostici non sono conclusivi o quando sono necessari risultati urgenti.
Le analisi eseguite su questi campioni includono il cariotipo (per esaminare la composizione cromosomica), la FISH (Ibridazione Fluorescente in Situ) e l'analisi cromosomica con microarray. In presenza di malformazioni fetali rilevate all’ecografia, il microarray è raccomandato poiché aumenta il tasso diagnostico rispetto al cariotipo standard. È importante notare che il cfDNA riflette il genoma placentare, quindi condizioni come il mosaicismo placentare confinato o raramente condizioni materne (es. neoplasie) possono spiegare risultati cfDNA discordanti con il cariotipo fetale.
Diagnosi Postnatale
In alcuni casi, la trisomia 21 o altre anomalie cromosomiche potrebbero non essere state diagnosticate in fase prenatale. Pertanto, la diagnosi viene effettuata dopo la nascita del bambino attraverso l'osservazione delle caratteristiche fisiche tipiche del neonato, come la forma del capo, il peso e le dimensioni ridotte rispetto alla media, e gli otto segni diagnostici (faccia piatta, displasia dell'orecchio, protrusione della lingua, angoli della bocca rivolti verso il basso, ipotonia, eccesso di pelle del collo, piega epicantica e ampio divario tra alluce e secondo dito del piede). Se sono presenti almeno sei di queste caratteristiche, la sindrome può essere stabilita con ragionevole certezza. La diagnosi clinica viene poi confermata in modo inequivocabile da un test del sangue chiamato esame del cariotipo, che esamina la composizione cromosomica del DNA del neonato, rivelando la presenza della copia extra del cromosoma 21.
Gestione e Percorsi di Cura: Una Vita Piena di Potenzialità
Vivere con la sindrome di Down oggi è una sfida piena di ostacoli che possono essere superati grazie a un approccio multidisciplinare e integrato alla cura e al supporto. Sebbene non esista una cura farmacologica per la sindrome di Down in sé, molti sintomi e problemi specifici possono essere trattati efficacemente, migliorando significativamente l'aspettativa e la qualità della vita delle persone affette.
Approccio Multidisciplinare e Interventi Precoci
L'obiettivo principale della gestione attuale è l'integrazione e il supporto delle persone con sindrome di Down, agevolandole nel condurre una vita piacevole e caratterizzata da tutte le occupazioni, dal lavoro allo studio e allo svago. Questo richiede un approccio sanitario che vada oltre i comuni controlli clinici, con la necessità di controlli specifici, finalizzati alla prevenzione o alla diagnosi precoce di quelle patologie che si possono presentare con una frequenza superiore alla norma. Trascurare queste patologie potrebbe limitare le potenzialità evolutive del bambino o far regredire le capacità dell’adulto.
Un ruolo cruciale è giocato dalla terapia riabilitativa precoce, che dovrebbe essere avviata fin dai primi mesi di vita. Questa terapia riguarda l'aspetto motorio, psico-motorio e logopedico, con lo scopo di agevolare lo sviluppo e l'apprendimento di abilità motorie e cognitive nei bambini. Un intervento precoce e mirato può fare una differenza sostanziale nello sviluppo delle abilità e nella gestione dei comportamenti, consentendo ai bambini di sviluppare le loro capacità, acquisirne di nuove in autonomia e favorire relazioni interpersonali soddisfacenti.
Il monitoraggio delle comorbilità è essenziale. Questo include:
- Cardiopatie: Controlli cardiologici regolari e, se necessario, interventi farmacologici o chirurgici.
- Patologie Tiroidee: Screening annuale per ipotiroidismo e altre disfunzioni tiroidee.
- Problemi Otorinolaringoiatrici e Oculari: Esami audiometrici regolari (dato l'alto rischio di perdita dell'udito e otiti ricorrenti) e controlli oculistici per cataratta, glaucoma e difetti refrattivi. L'uso di apparecchi acustici può essere di grande aiuto.
- Problemi Gastrointestinali: Monitoraggio per celiachia, malattia di Hirschsprung e altre anomalie digestive.
- Instabilità Vertebrale: Valutazione dell'instabilità atlanto-occipitale/assiale per prevenire la compressione del midollo spinale.
- Diabete e Obesità: Gestione dietetica e monitoraggio metabolico.
- Rischio Leucemie: Controlli ematologici regolari.
Inclusione Sociale e Qualità della Vita
L’inclusione sociale delle persone con sindrome di Down rappresenta oggi un obiettivo centrale nelle politiche educative, lavorative e sanitarie. Fortunatamente, i passi avanti sono tanti. Progetti efficaci promuovono l'autonomia e la partecipazione, sfidando cliché e comuni misconcezioni. È fondamentale utilizzare una terminologia rispettosa e aggiornata, evitando espressioni storiche e stigmatizzanti come "mongolismo" o "mongoloide".
L'obiettivo è consentire alle persone con sindrome di Down di realizzarsi pienamente, sia sul lavoro che nella vita privata. Spesso, non trovano le condizioni adatte per mettere a frutto il proprio potenziale a causa di barriere e una scarsa consapevolezza da parte della società riguardo all'inclusione e alle capacità individuali. Tuttavia, grazie al miglioramento dell'assistenza sanitaria e ai programmi di supporto, la maggior parte delle persone con sindrome di Down raggiunge e supera la soglia dei 60 anni di età, vivendo una vita sempre più ricca e significativa.
Aneuploidie ed anomalie cromosomiche
La Ricerca Scientifica e le Prospettive Future
La ricerca sulla sindrome di Down è estremamente attiva e multidisciplinare, esplorando diversi fronti per comprendere appieno questa condizione e migliorare le vite delle persone che ne sono affette. Gli studi attuali mirano a chiarire i meccanismi molecolari attraverso i quali l'anomalia genetica del cromosoma 21 esercita i suoi effetti complessi.
Le linee di ricerca più promettenti includono:
- Modulazione del Dosaggio Genico: Si studiano approcci per normalizzare l'espressione dei geni che si trovano sul cromosoma 21. Ad esempio, si mira a bersagliare geni specifici come DYRK1A, che si ritiene contribuiscano significativamente al fenotipo della sindrome di Down. Studi recenti hanno anche esplorato la possibilità di inattivare la terza copia del cromosoma 21 utilizzando geni specifici a RNA, come XIST.
- Controllo dell'Infiammazione e degli Interferoni: La sindrome di Down è associata a una disregolazione immunitaria e a una maggiore sensibilità alle infezioni, con un'alterata risposta infiammatoria. La ricerca si concentra su come modulare queste risposte per ridurre le comorbilità e migliorare la salute generale.
- Sostegno alla Bioenergetica Mitocondriale: Le alterazioni del metabolismo ossidativo e della funzione mitocondriale sono state identificate come processi chiave influenzati dal sovradosaggio genico. Interventi mirati a migliorare la funzione mitocondriale potrebbero avere un impatto positivo sulle manifestazioni neurologiche e su altri sistemi.
- Approcci di Epigenetic Editing: Si esplorano tecniche per modificare l'espressione genica senza alterare la sequenza del DNA, offrendo la possibilità di "silenziare" o modulare l'attività dei geni in eccesso.
- Ricerca di Nuovi Trattamenti per le Comorbilità: Oltre a una potenziale "cura", una parte significativa della ricerca è dedicata allo sviluppo di trattamenti più efficaci per le numerose comorbilità (cardiopatie, problemi gastrointestinali, problemi neurologici, ecc.) che affliggono le persone con sindrome di Down.
- Raffinemento Diagnostico: Continuano gli sforzi per rendere la diagnosi ancora più precisa, sia in fase prenatale che postnatale, e per sviluppare nuovi marker che possano aiutare a prevedere la gravità delle manifestazioni cliniche.
Storicamente, si è ipotizzata l'esistenza di una "regione critica della sindrome di Down" (Down Syndrome Critical Region, DSCR) sul cromosoma 21, la cui duplicazione si pensava fosse sufficiente a causare il fenotipo. Tuttavia, la visione attuale privilegia un modello poligenico, in cui più regioni e reti geniche interagiscono per produrre il fenotipo complessivo della sindrome. La comprensione di queste interazioni complesse è fondamentale per sviluppare interventi terapeutici mirati ed efficaci.
La ricerca si avvale anche di modelli preclinici, come animali di laboratorio, per studiare gli effetti di geni specifici e di gruppi di geni coinvolti nella sindrome, e per testare potenziali rimedi. Questi studi non solo mirano a una comprensione più profonda dei meccanismi patologici, ma cercano anche di identificare bersagli terapeutici per migliorare la qualità della vita e le potenzialità delle persone con sindrome di Down.
tags: #trisomia #21 #traslocazione #robertsoniana