La sindrome di Turner è una condizione genetica complessa che incide profondamente sullo sviluppo delle donne, manifestandosi con una gamma estremamente variabile di caratteristiche cliniche. Questa anomalia cromosomica, caratterizzata dalla mancanza totale o parziale di un cromosoma X, si verifica in circa 1 donna ogni 2000/2500 nate vive, configurando una condizione clinica peculiare. La possibilità di identificare precocemente indicatori di questa sindrome, anche in epoca prenatale, assume un ruolo cruciale per una gestione tempestiva e mirata. Tra questi indicatori, la translucenza nucale, sebbene non specifica, può rappresentare un segnale d'allarme che richiede ulteriori indagini diagnostiche, orientando i clinici verso un percorso di approfondimento volto a delineare il quadro genetico e clinico della paziente. La comprensione delle basi genetiche, delle manifestazioni fenotipiche e delle strategie diagnostiche e terapeutiche è fondamentale per offrire un supporto completo e migliorare la qualità della vita delle persone affette.
La Sindrome di Turner: Un'Anomalia Cromosomica del Sesso Femminile
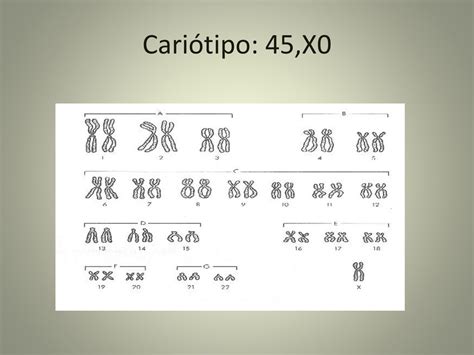
La sindrome di Turner è un'anomalia cromosomica che colpisce gli individui di sesso femminile, derivante dalla mancanza totale o parziale di un cromosoma X, una condizione nota come monosomia totale o parziale del cromosoma X. In termini più specifici, si fa riferimento a una aneuploidia, ovvero una variazione del numero di cromosomi. L'assetto cromosomico più comune che caratterizza la sindrome è il cariotipo 45,X0, dove manca un intero cromosoma X.
Tuttavia, il quadro genetico può essere più variegato, presentando diverse varianti. Tra queste si annovera il cariotipo 46,X,i(Xq), dove si forma un isocromosoma costituito da due braccia lunghe del cromosoma X. Un'altra variante include il cariotipo 46,XXq- o 46,XXp-, causato da delezioni del braccio lungo o corto del cromosoma X. Inoltre, esiste il cariotipo 46,X,r(X), che implica delezioni di porzioni sia del braccio lungo che del braccio corto del cromosoma X, portando alla formazione di un cromosoma X ad anello. Esistono anche forme a mosaico con vari cariotipi, come 45,X/46,XY o 45,X/47,XXX, dove solo alcune cellule presentano l'anomalia cromosomica, mentre altre hanno un assetto normale o diverso. Nelle ragazze con mosaicismo, il fenotipo può spaziare da quello della tipica sindrome di Turner a quello normale, con un grado di espressività correlato alla percentuale di cellule monosomiche.
Circa il 45% delle ragazze affette dalla sindrome di Turner ha un cariotipo 45,X, e si stima che circa l'80% di queste abbia perduto il cromosoma X paterno. La maggior parte del restante 55% presenta mosaicismo, con esempi come 45,X/46,XX oppure 45,X/47,XXX. È interessante notare che persone con un cromosoma X e un mosaicismo che coinvolge un cromosoma Y (ad esempio, alcune cellule con 46, XY) possono presentare un fenotipo maschile o femminile o persino genitali ambigui, rendendo la diagnosi e la gestione ancora più complesse. La delezione del braccio corto del cromosoma X sembra avere un ruolo fondamentale nella determinazione del fenotipo tipico della sindrome.
L'origine di questa anomalia cromosomica è solitamente attribuita alla non disgiunzione di un cromosoma sessuale durante la meiosi, più frequentemente nell'ovogenesi, che comporta la perdita di un cromosoma sessuale materno. L'unione di un gamete privo di cromosomi sessuali con un gamete normale contenente un cromosoma X dà origine a individui affetti da questa sindrome. Sebbene la sindrome di Turner sia una condizione rara, che si verifica in circa 1/2500 femmine nate vive in tutto il mondo, è importante sottolineare che il 99% dei concepimenti con cariotipo 45,X esita in un aborto spontaneo, evidenziando la gravità di questa anomalia cromosomica per lo sviluppo fetale.
Fenotipo e Manifestazioni Cliniche della Sindrome di Turner: Un Quadro Variabile
Il fenotipo della sindrome di Turner è notoriamente variabile e può essere espresso in modo diverso, dalle forme tipiche associate al cariotipo 45,X0 a forme che mostrano un fenotipo parziale, come quelle in mosaico. Questa variabilità è strettamente correlata all'epoca di insorgenza e alla specificità dell'anomalia cromosomica.
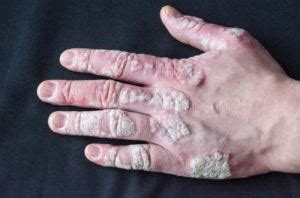
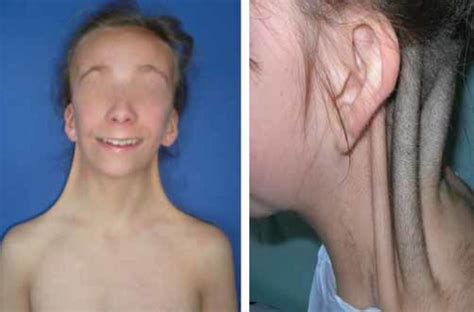
Manifestazioni in Epoca Prenatale e Neonatale:In un terzo dei casi, la presenza della sindrome può essere sospettata già in epoca prenatale o neonatale. Un indicatore significativo è l'igroma cistico, una malformazione linfatica che può essere rilevata ecograficamente. Oltre a ciò, possono manifestarsi cardiopatie congenite, come la coartazione aortica o il difetto interventricolare (DIV). Alla nascita e immediatamente dopo, i neonati sono solitamente interessati in modo molto lieve; tuttavia, alcuni presentano un marcato linfedema del dorso delle mani e dei piedi, oppure linfedema o pieghe cutanee nella parte posteriore del collo. Altre anomalie frequenti comprendono il collo palmato (pterigio del collo), una displasia ungueale, una rima buccale ampia con angoli rivolti in basso, e padiglioni auricolari displasici. La presenza di un soffio sistolico può indicare una cardiopatia congenita sottostante, come la coartazione aortica o il DIV.
Manifestazioni nell'Infanzia e nell'Adolescenza:Nella maggioranza dei casi, la diagnosi viene posta durante l'infanzia o l'adolescenza, principalmente a causa di due segni clinici preponderanti: la bassa statura e l'amenorrea primaria. Le ragazze affette sono spesso di statura inferiore rispetto ai familiari e l'obesità è un problema frequente. Altre caratteristiche fisiche includono un collo corto con pterigio, teletelia (un aumento della distanza tra i capezzoli), un'attaccatura bassa dei capelli sulla nuca, e un torace ristretto che può assumere l'aspetto di un "petto a scudo". Possono essere presenti numerosi nevi iperpigmentati. A livello scheletrico, si osserva spesso un cubito valgo (un aumento dell'angolo di valgismo nel gomito), l'accorciamento del quarto metacarpo e metatarso, dita prominenti con vortici nei dermatoglifi (le creste caratteristiche sui palmi che danno origine alle impronte digitali alle estremità delle dita) e l'ipoplasia delle unghie. Un segno radiologico peculiare è il segno di Kosowicz, che indica un aumento dell'angolo carpieno al polso e un aspetto ad incudine del bordo interno della tibia.
Manifestazioni alla Pubertà e in Età Adulta:La pubertà è un'epoca cruciale per la diagnosi della sindrome di Turner a causa della disgenesia gonadica. Questa si manifesta con amenorrea primaria (assenza di mestruazioni) e assenza di sviluppo dei caratteri sessuali secondari, come lo sviluppo del seno. Il quadro endocrino è caratterizzato da disgenesia gonadica, con un deficit di estrogeni e un aumento delle gonadotropine. Le ovaie sono estremamente ipoplasiche e assumono un aspetto fibroso, spesso descritte come "gonadi a bandelletta" (bande bilaterali di stroma fibroso e assenza di sviluppo di ovuli), causando insufficienza ovarica prematura. L'utero è tipicamente ipoplasico. In alcuni casi, può verificarsi agenesia ovarica monolaterale, spesso accompagnata dall'assenza, dallo stesso lato, di rene, uretere, tuba e metà dell'utero. Questa insufficienza ovarica precoce si traduce in amenorrea nella maggior parte delle pazienti; sebbene alcune possano avere un menarca, successivamente sviluppano amenorrea secondaria. È da notare che una piccola percentuale di pazienti può avere pubertà e funzione riproduttiva normali, sebbene la disgenesia gonadica si verifichi in quasi tutti i pazienti.
Indicatori Prenatali: Il Ruolo della Translucenza Nucale e Altri Segni Ecografici
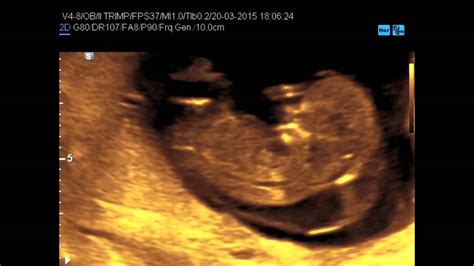
La diagnosi prenatale della sindrome di Turner rappresenta una sfida significativa, poiché non esistono segni ecografici specifici che, da soli, possano confermare la diagnosi. Tuttavia, l'ecografia di primo trimestre, in particolare l'analisi della translucenza nucale (TN), e la successiva ecografia morfologica, possono rivelare indicatori che, sebbene aspecifici, dovrebbero indurre a ulteriori accertamenti.
La translucenza nucale è la misurazione ecografica dello spessore dello spazio sottocutaneo tra la pelle e i tessuti molli che coprono la colonna cervicale del feto. Un aumento della translucenza nucale è un marker di screening consolidato per diverse anomalie cromosomiche, tra cui la trisomia 21 (sindrome di Down), e anche per la sindrome di Turner. Sebbene una translucenza nucale aumentata non sia diagnostica di per sé, la sua presenza in epoca prenatale, soprattutto se marcatamente elevata, è un forte campanello d'allarme che giustifica l'esecuzione di test diagnostici invasivi, come la villocentesi o l'amniocentesi, per l'analisi del cariotipo fetale.
Un segno ecografico prenatale di particolare rilevanza, spesso associato a un aumento della translucenza nucale nella sindrome di Turner, è l'igroma cistico. Questo si presenta come una malformazione del sistema linfatico, una sorta di sacca piena di liquido, solitamente localizzata nella regione nucale del feto. La presenza di igroma cistico, o di linfedema del dorso di piedi e mani, è un indicatore che può porre il sospetto della sindrome già in epoca prenatale o neonatale, a volte in un terzo dei casi. L'igroma cistico rappresenta una forma più grave di edema nucale e ha una forte associazione con la sindrome di Turner, sebbene possa essere riscontrato anche in altre anomalie cromosomiche o sindromi genetiche.
Oltre all'igroma cistico, altri reperti ecografici che possono suggerire una sindrome di Turner in epoca prenatale includono la presenza di cardiopatie congenite. Le malformazioni degli organi interni interessano, in particolar modo, l'apparato cardiovascolare. Le anomalie cardiache comuni rilevabili ecograficamente possono includere la coartazione aortica e il difetto interventricolare (DIV), che sono tra le più frequenti malformazioni associate alla sindrome.
La diagnosi ecografica prenatale, pur non essendo specifica, riveste un ruolo fondamentale nel percorso di screening. Sebbene "non vi siano segni specifici ecografici per la diagnosi di Sindrome di Turner", la rilevazione di questi marker non specifici - come l'aumento della translucenza nucale, l'igroma cistico o le anomalie cardiache - è cruciale per indirizzare i genitori verso esami diagnostici più approfonditi. La possibilità di porre diagnosi prenatale di sindrome di Turner esiste in alcuni casi, sin dal primo trimestre di gravidanza, specialmente quando si riscontrano tali anomalie.
La Dr.ssa Chiara Barone, medico specialista in Genetica Medica e cofondatrice di BGenetica, sottolinea l'importanza di questi screening. In questo contesto, anche il Cell-free DNA Screening for Sex Chromosome Aneuploidies by Non-Invasive Prenatal Testing (NIPT) in Maternal Plasma, come indicato in alcune ricerche, sta assumendo un ruolo crescente come metodo di screening non invasivo per le aneuploidie dei cromosomi sessuali, inclusa la sindrome di Turner, offrendo un'ulteriore opzione per l'identificazione precoce.
Diagnosi Post-Natale e Fisiopatologia Associata: Comprendere le Comorbilità
La diagnosi della sindrome di Turner, se non posta in epoca prenatale o neonatale, si basa sull'esame obiettivo e viene confermata dall'analisi citogenetica. Nei neonati, il sospetto può sorgere dalla presenza di linfedema marcato del dorso delle mani e dei piedi o di un collo palmato. In assenza di questi reperti evidenti, molte bambine vengono diagnosticate più tardi, durante l'infanzia o l'adolescenza, principalmente a causa di bassa statura, assenza di sviluppo puberale e amenorrea primaria.
La conferma diagnostica è ottenuta tramite test citogenetici, che includono il cariotipo standard, l'analisi dell'ibridazione fluorescente in situ (FISH) e/o l'analisi cromosomica con microarray. Questi esami consentono di identificare la monosomia X o le sue varianti a mosaico. È fondamentale, in tutti i pazienti con disgenesia gonadica, effettuare un'analisi citogenetica e uno studio con sonde specifiche per il cromosoma Y, al fine di escludere casi di mosaicismo con una linea cellulare che porta un cromosoma Y (ad esempio, 45,X/46,XY). Questi soggetti, pur avendo un fenotipo generalmente femminile che può presentare vari aspetti della sindrome di Turner, hanno un elevato rischio di sviluppare tumori delle gonadi, in particolare gonadoblastomi, alcuni dei quali possono diventare maligni. Per questo motivo, la rimozione profilattica della gonade è spesso raccomandata, sebbene rimanga un argomento controverso e soggetto a discussione clinica.
Fisiopatologia e Comorbilità:La sindrome di Turner è associata a una serie di anomalie multi-organo e deficit neurocognitivi, che richiedono un'attenta valutazione e gestione.
- Apparato Cardiovascolare: Le malformazioni degli organi interni interessano, in particolar modo, l’apparato cardiovascolare. Le comuni anomalie cardiache comprendono la coartazione aortica, la dilatazione aortica e la valvola aortica bicuspide. La coartazione dell'aorta associata può causare ipertensione arteriosa agli arti superiori, diminuzione dei polsi femorali e ridotta o assente pressione arteriosa nelle estremità inferiori. L'ipertensione si presenta spesso con l'avanzare dell'età anche in assenza di coartazione.
- Apparato Renale: Sono comuni malformazioni renali ed emangiomi. Queste possono variare da anomalie strutturali minori a condizioni più gravi che richiedono monitoraggio.
- Apparato Gastrointestinale: Occasionalmente, possono comparire telangectasie nel tratto gastrointestinale, con conseguente sanguinamento gastrointestinale o perdita proteica, che possono portare a complicazioni significative.
- Sistema Udito e Vista: Una perdita dell'udito può verificarsi, e strabismo e ipermetropia sono frequenti, aumentando il rischio di ambliopia.
- Malattie Autoimmuni ed Endocrine: Tiroidite, in particolare tiroidite di Hashimoto, diabete mellito e celiachia sono condizioni più frequenti rispetto alla popolazione generale, richiedendo uno screening e una gestione proattivi. Il quadro endocrino è dominato dalla disgenesia gonadica, con conseguente ipogonadismo ipergonadotropo e infertilità.
- Sistema Scheletrico: I neonati sono a più alto rischio di displasia dello sviluppo dell'anca. Circa il 10% degli adolescenti presenta scoliosi. L'osteoporosi e le fratture sono abbastanza frequenti tra le donne con sindrome di Turner, sottolineando l'importanza di un'adeguata supplementazione e monitoraggio della densità ossea.
- Funzione Riproduttiva: La disgenesia gonadica, caratterizzata dalle ovaie sostituite da bande bilaterali di stroma fibroso e dall'assenza di sviluppo di ovuli, si verifica in quasi tutti i pazienti. Sebbene tra il 15% e il 40% delle adolescenti con sindrome di Turner vada incontro a pubertà spontanea, solo dal 2 al 10% va incontro a menarca spontaneo, e la maggior parte delle pazienti è infertile a causa dell'insufficienza ovarica prematura. Le ovaie presentano tessuti connettivi con o senza follicoli, definiti "gonadi a bandelletta".
- Sviluppo Neurocognitivo: La disabilità intellettiva è rara, ma molte ragazze presentano disabilità dell'apprendimento non verbale, disturbo da deficit di attenzione/iperattività (ADHD) o entrambi. Conseguentemente, ottengono punteggi bassi nei test di performance e in matematica, anche se i loro punteggi nei componenti verbali dei test di intelligenza sono nella media o superiori. Questo suggerisce la necessità di un supporto educativo personalizzato.
Sindrome di Turner: cos'è, come si diagnostica e come si cura?
Strategie di Gestione e Supporto per la Sindrome di Turner
Non esiste un trattamento specifico per la condizione genetica sottostante della sindrome di Turner, in quanto essendo una malattia genetica, non esistono cure per la causa principale. Tuttavia, la gestione della sindrome è di tipo multidisciplinare e mira a trattare le manifestazioni cliniche, prevenire le complicanze e migliorare la qualità della vita delle pazienti. La gestione appropriata della sindrome di Turner varia in base all'età e comporta un'assistenza coordinata da parte di un team specialistico.
Gestione delle Comorbilità:La gestione completa deve includere lo screening e la gestione regolari di ipertensione, dislipidemia, intolleranza al glucosio e disfunzione tiroidea. È raccomandato un regolare follow-up con un team multidisciplinare che includa cardiologi, nefrologi, endocrinologi, audiologi, oftalmologi e ortopedici. Alcune visite di routine sono essenziali per identificare le problematiche associate:
- Valutazione cardiologica: Con ecocardiografia ed ECG al momento della diagnosi e valutazione continuata per monitorare le anomalie di conduzione e la dilatazione aortica. La coartazione aortica viene in genere riparata chirurgicamente, e altre anomalie cardiache sono monitorate e trattate secondo le necessità.
- Monitoraggio metabolico: Screening di routine per dislipidemia e ipertensione.
- Valutazione renale: Ecografia renale al momento della diagnosi, analisi delle urine annuale, azotemia e creatinina per i pazienti con anomalie del sistema renale.
- Udito e vista: Valutazione dell'udito da parte di un audiologo e audiogramma ogni 3 o 5 anni. Valutazione oftalmologica all'età di 12-18 mesi o al momento della diagnosi, e successivamente annuale.
- Sistema scheletrico: Valutazione annuale per la scoliosi/cifosi durante l'infanzia e l'adolescenza e valutazione per la lussazione delle anche.
- Funzione tiroidea: Test della funzione tiroidea al momento della diagnosi e successivamente ogni anno.
- Celiachia: Esame per la celiachia (ad esempio, i livelli di anticorpi endomisio).
- Diabete: Screening annuale per l'intolleranza al glucosio a partire dall'età di 10 anni.
- Crescita e sviluppo puberale: Stretto monitoraggio della crescita e dello sviluppo puberale.
Terapie Ormonali:
- Ormone della crescita: Il trattamento con l'ormone della crescita è in grado di stimolare la crescita, permettendo a molte ragazze di raggiungere una statura più vicina alla media. Questo può essere somministrato in combinazione con la terapia estrogenica fino a che le epifisi non sono fuse.
- Terapia sostitutiva degli estrogeni: La terapia sostitutiva degli estrogeni è in genere necessaria per avviare la pubertà ed è solitamente somministrata all'età di 12-13 anni. In seguito, vengono somministrati contraccettivi estroprogestinici per mantenere i caratteri sessuali secondari e la salute ossea. La continuazione della terapia con estrogeni aiuta a stabilire un'ottimale densità ossea e lo sviluppo scheletrico, riducendo il rischio di osteoporosi e fratture. Inoltre, la terapia con estrogeni può anche migliorare la capacità della ragazza di pianificare i compiti, prestare attenzione e valutare le relazioni visive e spaziali, influenzando positivamente le capacità neurocognitive.
Gestione del Linfedema:Il linfedema, una delle manifestazioni più comuni e visibili, solitamente può essere controllato con calze elastiche e altre tecniche come il massaggio per migliorare la circolazione linfatica e ridurre l'accumulo eccessivo.
Supporto Psicosociale e Fertilità:Non va sottovalutato l'aspetto psicologico delle pazienti affette dalla sindrome di Turner. Per molte donne, la sterilità e le caratteristiche fenotipiche peculiari della malattia potrebbero essere rigettate o divenire causa di malcontento, ansia e depressione. Fornire supporto psicosociale, neurocognitivo, educativo e basato sulla famiglia è fondamentale per migliorare la qualità della vita dei pazienti e dei loro familiari. Può essere utile sostenere ed aiutare queste donne mediante l'intervento di psicologi e mediante un approccio olistico alle pazienti, in ogni fase della malattia, da parte di tutti i componenti dell'équipe.
Le donne con sindrome di Turner a mosaico possono rimanere incinte e devono usare la contraccezione se sono sessualmente attive e desiderano prevenire una gravidanza. Tuttavia, la fertilità può essere limitata, quindi se desiderano una gravidanza, di solito richiedono interventi di fertilità alternativi, come la fecondazione in vitro con ovodonazione. L'assistenza alle persone con sindrome di Turner e alle loro famiglie deve comprendere anche una consulenza genetica approfondita per informare sulle implicazioni riproduttive e sui rischi di trasmissione.
tags: #translucenza #nucale #sindrome #di #turner