La sordità o ipoacusia congenita, ovvero quella condizione di deficit uditivo presente già alla nascita, rappresenta una sfida clinica e sociale di primaria importanza. Con un’incidenza di circa 1 bambino ogni 1.000 nati, questa patologia può manifestarsi con gradi diversi, da moderati a profondi, e si caratterizza solitamente per essere una condizione non progressiva. La comprensione di tale fenomeno richiede un'analisi multidisciplinare che integra genetica, neonatologia e otorinolaringoiatria.
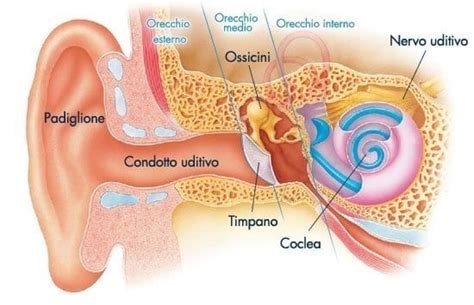
L'anatomia dell'udito e la classificazione delle perdite uditive
L'orecchio è un organo complesso suddiviso in tre parti principali, ciascuna con funzioni specifiche che contribuiscono al processo dell’udito: esterna, media e interna. L’orecchio esterno comprende il padiglione auricolare e il condotto uditivo, strutture che raccolgono e convogliano le onde sonore verso il timpano, dove le vibrazioni sonore sono trasformate in oscillazioni meccaniche. L’orecchio medio, separato dall’esterno proprio dal timpano, ospita una catena di ossicini (martello, incudine e staffa) che amplificano e trasmettono queste vibrazioni alla finestra ovale, una membrana che comunica con l’orecchio interno. Nell’orecchio interno, la coclea, una struttura a forma di spirale riempita di fluido, converte le vibrazioni meccaniche in segnali elettrici grazie al movimento delle cellule ciliate situate nella membrana basale. Queste cellule rispondono a specifiche frequenze sonore e trasmettono i segnali al nervo uditivo, che li invia al cervello.
Quando vi è un’alterazione della capacità di trasmissione delle onde sonore attraverso l’orecchio esterno e/o medio, si parla di disturbo di trasmissione; se invece il problema interessa l’orecchio interno, di solito la coclea, si parla di disturbo neurosensoriale. Questa forma è di solito irreversibile.
L'eziologia genetica: il ruolo fondamentale del DNA
Mentre fino a qualche anno fa si riteneva che le cause genetiche rappresentassero un terzo dei casi di ipoacusia nel bambino, attualmente grazie allo sviluppo delle diagnosi molecolari è stato possibile ricollegare a una causa genetica la maggioranza dei casi sporadici di ipoacusia. Oggi si stima che circa il 75% delle ipoacusie congenite sia riferibile a cause genetiche. Di queste, il 30% circa sono forme sindromiche, mentre il 70% sono forme non sindromiche, in cui la perdita dell’udito è l’unico sintomo presente.
Il gene connessina 26 (Cx26, indicato anche con la sigla GJB2), localizzato sul braccio lungo del cromosoma 13 (13q11), è il responsabile della maggior parte dei casi di sordità autosomica recessiva. In Italia, questo gene copre l’80% dei casi. Le connessine sono una famiglia di proteine presenti sulla membrana plasmatica che formano dei canali necessari per gli scambi e le comunicazioni tra le cellule. La mutazione 35delG nel gene GJB2 è in grado di spiegare il 50% di tutte le sordità genetiche. Accanto a questo, il gene GJB6 (connessina 30) gioca un ruolo cruciale: la contemporanea presenza nello stesso soggetto di una mutazione nel gene GJB6 e di una mutazione nel gene GJB2 causa ipoacusia congenita.
Malattie autosomiche dominanti e recessive
Meccanismi di trasmissione ereditaria
Le ipoacusie genetiche sono, nella maggior parte dei casi, malattie monogeniche. Il modello di ereditarietà autosomica recessiva è il più frequente: due copie del gene anomalo causano la malattia. Gli individui che ereditano un solo gene anomalo e un gene normale sono indicati come vettori (portatori sani) e non sono colpiti dalla malattia. Poiché si tratta di una patologia recessiva, i portatori non presentano alcuna manifestazione, ma in presenza di entrambi i partner eterozigoti, esiste un rischio del 25% di avere figli non udenti.
Esistono tuttavia altre modalità di trasmissione:
- Ereditarietà legata al cromosoma X (X-linked): Il gene coinvolto è localizzato sul cromosoma sessuale X. Poiché i maschi hanno un solo cromosoma X (XY), se ereditano il cromosoma con la mutazione svilupperanno la sordità. Le femmine, avendo due cromosomi X (XX), possono compensare una copia mutata con una normale, risultando portatrici sane. Esempi includono la sindrome di Norrie e la sindrome di Alport.
- Ereditarietà mitocondriale: Il genoma mitocondriale è ereditato esclusivamente per via materna. Una madre con una mutazione mitocondriale può trasmetterla a tutta la prole. Queste forme sono spesso associate a una elevata suscettibilità all'ototossicità da aminoglicosidi.
- Ereditarietà autosomica dominante: È sufficiente una singola copia del gene mutato per manifestare il disturbo. La sindrome di Waardenburg è l’esempio più comune, caratterizzata non solo da ipoacusia, ma anche da alterazioni pigmentarie (come un ciuffo bianco o occhi di colore diverso).
Diagnosi precoce e screening neonatale
L'importanza di una diagnosi tempestiva non può essere sottovalutata. "Individuare precocemente un sordo consente una precoce terapia", sottolinea Stefano Martinelli, direttore di neonatologia. In Italia, lo screening uditivo neonatale è garantito dai Livelli Essenziali di Assistenza (LEA). Il test delle otoemissioni acustiche permette di registrare i suoni emessi dalla coclea durante il sonno. Tuttavia, la copertura dello screening rimane insufficiente, con migliaia di bambini che ogni anno rischiano di non essere sottoposti al controllo necessario prima della dimissione ospedaliera.
I segnali d’allarme nei primi mesi di vita, come la mancata reazione ai suoni o un ritardo nello sviluppo del linguaggio, sono spesso sottovalutati dai genitori o interpretati erroneamente. È vitale che, in presenza di dubbi, si proceda a una valutazione otorinolaringoiatrica esperta.
Analisi genetica e consulenza riproduttiva
Le indagini genetiche per identificare le mutazioni possono individuare circa il 95% delle mutazioni note. Effettuare lo screening sui genitori permette di identificare le coppie a rischio di avere un figlio affetto da patologie genetiche e di pianificare, se necessario, una diagnosi prenatale.
Se entrambi i genitori sono portatori della mutazione 35delG, il rischio di avere un figlio affetto da sordità neurosensoriale bilaterale profonda è del 25%. È importante sottolineare che la negatività al test per la mutazione 35delG non esclude la presenza di altre mutazioni rare nel medesimo gene o in altri loci. Per questo, la consulenza genetica pre-test è un passaggio obbligato per interpretare correttamente le probabilità e i limiti delle indagini molecolari disponibili oggi.
Verso una gestione multidisciplinare
La gestione del bambino sordo richiede un approccio che superi le carenze organizzative. La creazione di centri di riferimento regionali e la formazione di personale specializzato sono essenziali. Le opportunità di intervento moderno, come l’applicazione di protesi acustiche e, nei casi più gravi, l’impianto cocleare bilaterale, hanno rivoluzionato la prognosi di molti bambini. Tuttavia, l'attenzione deve rimanere alta anche verso le cause ambientali: infezioni prenatali (come citomegalovirus o rosolia), esposizione a farmaci ototossici o infezioni post-natali (meningite, morbillo) restano fattori critici che devono essere monitorati costantemente per garantire un percorso di crescita armonico e inclusivo.