La sindrome di Klinefelter è una condizione genetica che colpisce i maschi, caratterizzata dalla presenza di un cromosoma X in più nell'assetto genetico. Normalmente, i maschi hanno un cromosoma X e uno Y (XY), mentre le femmine hanno due cromosomi X (XX). Nei soggetti con sindrome di Klinefelter, l'assetto cromosomico tipico è 47,XXY, il che significa che presentano due cromosomi X e un cromosoma Y, anziché uno X e uno Y. Questa anomalia cromosomica, di natura non ereditaria, insorge a causa di un errore durante la divisione cellulare (meiosi) nella formazione dell'ovocita o dello spermatozoo.
La Diagnosi Prenatale: Amniocentesi e Test Genetici
La possibilità di diagnosticare la sindrome di Klinefelter durante la gravidanza è diventata sempre più concreta grazie ai progressi della medicina. Il test del DNA fetale circolante (NIPT - Non-Invasive Prenatal Testing) rappresenta un primo screening che, in caso di risultato positivo, necessita di essere confermato da analisi genetiche più invasive. Tra queste, l'amniocentesi riveste un ruolo cruciale. L'amniocentesi consiste nel prelievo di una piccola quantità di liquido amniotico, che circonda il feto, per analizzarne il contenuto genetico.
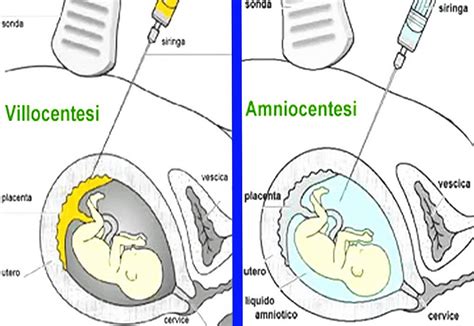
È fondamentale sottolineare che il NIPT è un test di screening e non diagnostico. Ciò significa che un risultato positivo non conferma inequivocabilmente la presenza della sindrome nel feto. Sussiste infatti la possibilità di un "falso positivo", in cui l'anomalia cromosomica è presente solo nella placenta e non nel feto stesso, oppure di una discrepanza tra il sospetto del NIPT e la reale costituzione cromosomica fetale. Per questo motivo, in caso di esito sospetto del NIPT, è indispensabile procedere con un'indagine più approfondita come l'amniocentesi o la villocentesi (prelievo dei villi coriali), che permettono di eseguire il cariotipo tradizionale e ottenere una diagnosi citogenetica certa. La comunicazione di una diagnosi prenatale, essendo spesso inaspettata, può generare forte preoccupazione nei futuri genitori, rendendo essenziale un supporto medico e psicologico adeguato.
Manifestazioni Cliniche della Sindrome di Klinefelter: Una Panoramica per Età
Le manifestazioni cliniche della sindrome di Klinefelter variano significativamente da individuo a individuo, influenzate dall'assetto del cariotipo specifico e dall'età del paziente.
Epoca Prenatale
Come accennato, la diagnosi prenatale sta acquisendo sempre maggiore rilevanza. L'identificazione dell'anomalia cromosomica può avvenire tramite procedure invasive come l'amniocentesi o la villocentesi, con l'esecuzione del cariotipo tradizionale. Il test del DNA fetale circolante (NIPT) può sospettare la condizione, ma richiede sempre una conferma diagnostica.
Età Pediatrica
Nel caso in cui la diagnosi non sia stata effettuata in epoca prenatale, il riconoscimento della sindrome in età pediatrica può essere più complesso, dato che i segni e i sintomi sono ancora poco specifici. I bambini affetti da sindrome di Klinefelter non presentano solitamente caratteristiche facciali particolari o disturbi significativi della crescita. In una percentuale di casi, si può tuttavia osservare un ritardo nell'acquisizione delle tappe dello sviluppo motorio, manifestato da ipotonia. Più frequentemente, si riscontra un ritardo nell'acquisizione delle fasi dello sviluppo del linguaggio, con una ridotta abilità espressiva ma una buona capacità di comunicazione non verbale. In queste circostanze, è raccomandato un consulto con il servizio di neuropsichiatria infantile, concordato con il pediatra curante, per una valutazione completa delle abilità del bambino e per fornire il necessario supporto ai genitori.

Adolescenza: La Comunicazione della Diagnosi
Durante l'adolescenza, i segni e i sintomi della sindrome di Klinefelter tendono a diventare più evidenti. Il pediatra, nel monitorare lo sviluppo puberale, deve prestare particolare attenzione alle modifiche del volume testicolare. Un sospetto clinico in questa fase impone l'invio al Centro di riferimento per la valutazione del cariotipo. Per i ragazzi, sia con diagnosi nota che non ancora diagnosticata, questo periodo di crescita fisica e psicologica può essere caratterizzato da difficoltà comportamentali e relazionali. È opportuno, in questi casi, considerare un nuovo contatto con il servizio di neuropsichiatria infantile. Un aspetto cruciale in questa fase è la comunicazione della diagnosi al ragazzo, un percorso che deve essere attentamente pianificato e supportato dall'equipe medica, in particolare dall'endocrinologo pediatra, per l'eventuale impostazione della terapia con testosterone.
La Klinefelter nell'adolescenza e adulto (aspetto psicologico)
Età Adulta: Gestione e Complicanze
In età adulta, nonostante i segni e i sintomi siano solitamente più marcati, la sindrome di Klinefelter può ancora passare inosservata. È pertanto fondamentale ricercare i segni e i sintomi specifici per indirizzare il soggetto verso un Centro di riferimento per una presa in carico multidisciplinare. Le due caratteristiche cliniche predominanti in età adulta sono l'ipogonadismo ipergonadotropo (una ridotta produzione di testosterone) e l'infertilità, spesso associata ad azoospermia (assenza di spermatozoi nell'eiaculato).
I livelli di testosterone risultano inferiori alla norma nel 65-85% dei casi. Lo specialista endocrinologo valuterà l'opportunità e le modalità di prescrizione della terapia sostitutiva, monitorando al contempo le possibili comorbidità associate alla sindrome, quali disturbi metabolici, cardiovascolari, ossei, neoplastici e autoimmuni. Altri segni e sintomi che possono manifestarsi includono una statura generalmente elevata, adiposità addominale, ginecomastia (sviluppo delle ghiandole mammarie nell'uomo, presente solo nel 10% dei casi) e osteoporosi.
La gestione della fertilità in età adulta prevede una consulenza andrologica e genetica. Sebbene la quasi totalità degli individui con sindrome di Klinefelter non produca spermatozoi in modo efficace, in alcuni casi vi è la possibilità che all'interno del testicolo vi siano zone in cui si sono sviluppati spermatozoi, senza tuttavia riuscire ad esserne espulsi. Le tecniche di fecondazione assistita, come la TESE (testicular sperm extraction), possono offrire una speranza in questi casi, ma richiedono un'attenta valutazione preliminare.
Aspetti Genetici e Meccanismi della Sindrome
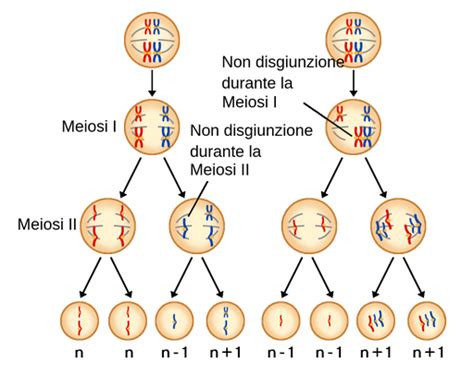
La sindrome di Klinefelter è causata dalla presenza di almeno un cromosoma X soprannumerario in un individuo di sesso maschile. L'anomalia cromosomica deriva da un evento di non disgiunzione dei cromosomi durante la meiosi. Questo può verificarsi:
- Origine paterna: Durante la meiosi I, i cromosomi sessuali paterni (X e Y) non si separano correttamente, portando alla formazione di uno spermatozoo con due cromosomi (XY). La fecondazione di un ovulo femminile (X) darà origine a un embrione XXY.
- Origine materna: Durante la meiosi II, i cromatidi fratelli di uno dei cromosomi X materni non si separano, generando un ovulo con due cromosomi X (XX). La fecondazione con uno spermatozoo Y produrrà un embrione XXY. Si ritiene che la trasmissione di origine materna sia più frequente.
Nei mammiferi con più cromosomi X, uno di essi viene inattivato per compensare la dose genica. Questo fenomeno, noto come inattivazione del cromosoma X o lyonizzazione, si verifica anche nei maschi XXY, sebbene non elimini completamente gli effetti dell'X soprannumerario. Il gene XIST, situato sul cromosoma X, gioca un ruolo chiave in questo processo.
Varianti della Sindrome di Klinefelter e Manifestazioni Specifiche
Oltre al cariotipo più comune 47,XXY, esistono varianti della sindrome di Klinefelter, caratterizzate da un numero maggiore di cromosomi X o Y:
- 48,XXYY: Si verifica in circa 1 caso ogni 18.000-40.000 nascite maschili. Il fenotipo è simile al 47,XXY, ma con una maggiore altezza media in età adulta.
- 49,XXXXY: Una rara polisomia che si riscontra in circa un caso ogni 85.000 bambini maschi. Questa condizione è generalmente riconosciuta precocemente a causa di deficit gravi, tra cui ritardo mentale marcato, dismorfismi facciali, criptorchidismo, genitali ambigui e difetti scheletrici e cardiaci.
- Varianti più rare: Sono stati riportati casi di cariotipi come 48,XXXY, 48,XXYY, 49,XXYYY, 48,XYYY, 49,XYYYY, e 49,XXYYY, con quadri fenotipici che variano da meno gravi a più severi rispetto al cariotipo classico.
In alcuni individui con sindrome di Klinefelter, si può osservare anche un mosaicismo nel cariotipo, come la forma 47,XXY/46,XY. Questo comporta gradi variabili di insufficiente spermatogenesi e un ampio spettro di manifestazioni cliniche.
Impatto Cognitivo e Comportamentale
Sebbene il ritardo mentale sia presente solo nel 10% dei soggetti con sindrome di Klinefelter, i problemi cognitivi tendono ad essere più selettivi e meno pervasivi rispetto ad altre sindromi da polisomia del cromosoma X. Sul piano neurologico, la sindrome è associata a un ridotto sviluppo del linguaggio, con difficoltà nell'espressività, anomia (difficoltà nel trovare le parole) e disartria.
L'intelligenza generale, misurata dal QI totale, rientra nei limiti di normalità. Tuttavia, si osserva spesso una discrepanza tra il QI verbale e il QI di performance, con un deficit più frequente a carico delle abilità verbali, evidente soprattutto durante il periodo scolare. I ragazzi affetti possono manifestare problemi di lettura, articolazione delle parole e scrittura, con difficoltà matematiche che emergono successivamente.
Nell'adolescenza, possono comparire ridotta fiducia in sé stessi, riservatezza, e difficoltà nel contenere gli impulsi e nell'accettare le regole. Questi sintomi possono avere implicazioni a lungo termine sulla socialità e sul rendimento scolastico, persistendo talvolta in età adulta.

Caratteristiche Fisiche e Segni Associati
Le caratteristiche fisiche associate alla sindrome di Klinefelter possono variare ampiamente. Tra le più comuni si annoverano:
- Statura elevata: Le gambe e le braccia possono essere più lunghe del normale.
- Scarsa muscolatura e ridotta peluria corporea.
- Spalle strette e fianchi larghi.
- Ginecomastia: Sviluppo delle ghiandole mammarie (nel 10% dei casi).
- Testicoli piccoli: Questa è una caratteristica quasi costante, presente a tutte le età per ipoplasia delle cellule germinali e interstiziali.
- Voce acuta.
- Rapporto tra indice e anulare (digit ratio 2D:4D) maggiore: Questo rapporto, più simile a quello femminile, è legato ai bassi livelli di testosterone prenatale.
Inoltre, vi è un aumentato rischio di osteoporosi, disordini autoimmuni della tiroide e diabete mellito di tipo 2. Alcuni studi hanno associato la sindrome a un'altezza leggermente maggiore della media e a una predisposizione al sovrappeso.
Gestione Medica e Supporto
La sindrome di Klinefelter è una condizione cronica e invalidante che richiede un approccio assistenziale multidisciplinare. Le cure mediche specialistiche includono:
- Terapia sostitutiva di testosterone: Circa due individui su tre affetti da sindrome di Klinefelter necessitano di questa terapia per contrastare i disturbi associati alla carenza di questo ormone. La terapia può iniziare in adolescenza e proseguire per tutta la vita adulta.
- Monitoraggio delle comorbidità: L'endocrinologo deve monitorare le possibili complicanze metaboliche, cardiovascolari, ossee, neoplastiche e autoimmuni.
- Supporto nella comunicazione della diagnosi: Particolarmente importante nelle fasi pre-puberale e adolescenziale.
- Supporto psicologico: L'intervento psicologico è una risorsa fondamentale per sostenere il soggetto, la sua famiglia e le coppie durante il percorso diagnostico e terapeutico.
- Valutazione andrologica e genetica: Essenziali per la gestione della fertilità e per la consulenza in caso di desiderio di procreazione.
La sindrome di Klinefelter è inserita tra le patologie croniche e invalidanti nei Livelli Essenziali di Assistenza (LEA) italiani, con un codice specifico che ne facilita l'accesso alle cure e alle esenzioni.
Sotto-diagnosi e Consapevolezza Pubblica
Nonostante la sindrome di Klinefelter non sia rara (si stima 1 maschio affetto ogni 500-1000 nati), è ancora ampiamente sotto-diagnosticata. Si stima che solo il 25-30% delle persone affette sappia di avere questa sindrome. Questa carenza diagnostica, unita alla complessità delle manifestazioni cliniche, contribuisce a una scarsa consapevolezza pubblica. Anche se ampiamente studiata in ambito medico, la sindrome è quasi sconosciuta alla popolazione generale, e ogni diagnosi suscita spesso reazioni drammatiche e allarmistiche.
Per avere informazioni sulla sindrome di Klinefelter, conoscere i centri clinici di diagnosi e cura e le associazioni di pazienti, è possibile rivolgersi al Telefono Verde Malattie Rare (TVMR) al numero 800.89.69.49.
È importante ricordare che le informazioni fornite in questo articolo non sostituiscono il parere medico e non sono da considerarsi consigli medici.