La sindrome di Down (SD), o trisomia 21, è una condizione genetica causata dalla presenza di materiale cromosomico in eccesso del cromosoma 21. Si manifesta quando, prima o dopo il concepimento, si verificano anomalie durante il meccanismo di separazione dei cromosomi. Questo evento è spontaneo, e sebbene l'età materna avanzata sia un fattore di rischio riconosciuto, la sindrome può interessare gravidanze in donne di qualsiasi età. Le paure e i dubbi, come quelli di Mina, che riflette sull'esperienza della sorella, attraversano la mente di tanti futuri genitori, rendendo fondamentale un'informazione chiara e aggiornata. È essenziale, infatti, utilizzare una terminologia rispettosa, evitando espressioni storiche come "mongolismo" o "mongoloide", che sono inappropriate e stigmatizzanti.
Comprendere la Sindrome di Down: Una Panoramica Genetica
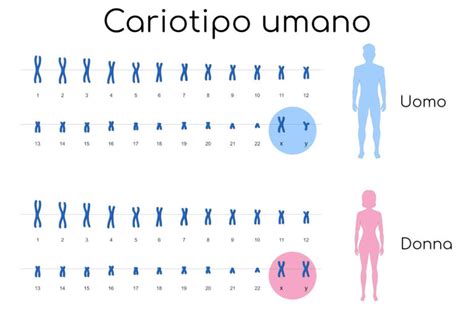
Normalmente, ogni cellula umana contiene 46 cromosomi, organizzati in 23 coppie. La sindrome di Down è caratterizzata dalla presenza di una copia supplementare del cromosoma 21, il cromosoma umano più piccolo. Questa anomalia cromosomica è definita trisomia 21 e ne esistono diverse tipologie, che influenzano anche il rischio di ricorrenza in gravidanze successive.
Le Tipologie di Trisomia 21
La trisomia 21 si presenta principalmente in tre forme:
Trisomia 21 Libera (circa 95% dei casi): Questa è la forma più comune e si verifica quando il cromosoma 21 è presente tre volte in tutte le cellule. L'origine è un errore casuale durante la divisione cellulare dei gameti (ovuli o spermatozoi), fenomeno chiamato "non disgiunzione". Nella maggior parte dei casi, l'errore avviene durante la meiosi I materna, ma una quota minoritaria (circa 5-10%) è di origine paterna. Questo tipo di trisomia non è ereditario nel senso stretto, ma il rischio aumenta con l'età materna.
Sindrome di Down da Traslocazione Robertsoniana (circa 3-4% dei casi): In questa forma, il cromosoma 21 supplementare, o meglio una sua parte (il braccio lungo), si attacca a un altro cromosoma acrocentrico, più spesso il 14 o il 15, e più raramente a un altro cromosoma 21. Quando non si ha né perdita né aggiunta di materiale genetico, si parla di traslocazione bilanciata, e un genitore può esserne portatore senza manifestare la sindrome. Questa condizione, a differenza della trisomia libera, può essere ereditata. I cromosomi acrocentrici hanno bracci corti che contengono principalmente regioni organizzatrici del nucleolo (rDNA), che sono ridondanti nel genoma umano.
Mosaicismo (1-2% dei casi): Questa forma, di più raro riscontro, è caratterizzata dalla contemporanea presenza, in percentuali diverse da individuo a individuo, di due tipi di cellule: alcune con un normale corredo cromosomico (46 cromosomi) e altre con 47 cromosomi, inclusa la copia extra del cromosoma 21. Il quadro clinico può essere più sfumato in funzione dell'entità del mosaicismo, ovvero della prevalenza dell'uno o dell'altro tipo di cellule.
I geni, segmenti di acido desossiribonucleico (DNA), contengono le istruzioni che determinano l'aspetto e il funzionamento dell'organismo. La presenza di un cromosoma supplementare modifica l'equilibrio genetico, portando a un variabile grado di ritardo nello sviluppo mentale, fisico e motorio.
Fattori di Rischio e Incidenza della Sindrome di Down
Le cause precise che determinano l'insorgenza della sindrome di Down sono ancora sconosciute, ma sono stati identificati diversi fattori di rischio.
Età Materna: Un Fattore Predisponente Cruciale
L'aumento dell'età materna è il principale fattore associato alla trisomia 21 da non-disgiunzione. Questo errore meiotico, per lo più di origine materna, risulta più frequente a mano a mano che l'età della donna è più elevata. Nonostante ciò, circa l'80% dei nati con sindrome di Down riguarda gravidanze in donne di età inferiore ai 35 anni. Questo dato è spiegato dal fatto che, complessivamente, in questa fascia di età si concepiscono e nascono più bambini.
L'Influenza dell'Età Paterna
Sebbene meno approfondita e di influenza inferiore (circa 5-10% dei casi), esiste anche un fattore legato all'età paterna. Raramente, una piccola quota di non-disgiunzioni può essere di origine paterna.
Traslocazione del Cromosoma 21 e Ereditarietà
La sindrome di Down è ereditaria solo nel caso in cui uno dei due genitori sia portatore di una traslocazione bilanciata. Se la madre è portatrice di traslocazione, c'è un rischio del 10-15% in più di avere un bambino con sindrome di Down; se l'origine è paterna, il rischio è di circa l'1-3%. Questo rischio non è collegato all'età. È indicato il cariotipo dei genitori in caso di trisomia 21 da traslocazione Robertsoniana per valutare il rischio di ricorrenza.
Altri Possibili Fattori
Alcuni studi hanno esplorato altri potenziali fattori, sebbene con evidenze meno consolidate:
- Fumo e consumo di prodotti a base di nicotina.
- Consumo di alcolici: può ridurre nel tempo l'assorbimento di vitamina B12 e acido folico, specialmente in presenza di una mutazione del gene MTHFR, che regola il metabolismo dei folati.
- Status socio-economico: uno studio condotto in diversi stati americani ha osservato che la bassa scolarizzazione familiare e uno status economico precario possono incidere sul maggiore tasso di Trisomia 21. Ciò è probabilmente dovuto a un accesso più complicato ai programmi sanitari di screening e una minore consapevolezza verso uno stile di vita più salutare.
Prevalenza e Distribuzione
A livello globale, la Sindrome di Down si verifica in un caso su 1.000-1.200 nati vivi, anche se la prevalenza alla nascita in un determinato Paese dipende ampiamente da fattori non medici, come le politiche pubbliche in materia di diagnosi prenatale e assistenza ai disabili, e dall'opinione pubblica riguardo alla malattia e all'aborto, con variazioni tra 1/400 e 1/3.000 nati vivi. La sindrome di Down è presente tra le diverse etnie senza alcuna distinzione, a variare è piuttosto la possibilità di accesso all'assistenza pre e post-natale, che dipende dalle condizioni sociali ed economiche delle diverse aree geografiche.

Caratteristiche Cliniche e Aspetti dello Sviluppo nella Sindrome di Down
La sindrome di Down colpisce diverse parti del corpo, e i segni clinici comprendono disabilità intellettiva di grado variabile, ipotonia muscolare e lassità articolare quasi sempre presenti, alterazioni morfologiche e un aumento del rischio di complicanze mediche.
Aspetti Fisici Comuni
I tratti fisici, anche se ogni persona è unica e può presentarli in misura variabile o non presentarli affatto, possono includere:
- Testa e Volto: Il cranio è di norma piccolo e potrebbe presentare un appiattimento a livello occipitale. Le fontanelle, nei neonati, sono larghe e si chiudono in ritardo rispetto alla norma. Il viso è spesso largo e piatto con un naso piccolo. Gli occhi hanno un tipico taglio inclinato verso l'alto e la pelle della palpebra superiore copre l'angolo interno dell'occhio (plica epicantale). A volte, la lingua è grande, e spesso la lingua più grande e il tono muscolare del volto ridotto fanno sì che i bambini tengano la bocca aperta. Le orecchie sono piccole e arrotondate e hanno un impianto basso nella testa.
- Collo: Il collo è spesso tozzo ed è presente una plica laterale (pterigio), talvolta con un eccesso di cute nella parte posteriore.
- Mani e Dita: Le mani sono usualmente piccole con dita brevi; è di frequente rilievo una plica palmare unica bilaterale. Le dita sono corte e il dito mignolo, che spesso ha 2 falangi invece di 3, è curvato verso l'interno.
- Corpo: I neonati con sindrome di Down tendono a essere di bassa statura e presentano un rischio aumentato di sviluppare obesità. L'ipotonia muscolare e la lassità articolare sono quasi sempre presenti.
È importante notare che alcuni neonati non presentano tratti del viso caratteristici evidenti alla nascita e li sviluppano invece durante la prima infanzia.

Sviluppo Cognitivo e Comportamentale
La sindrome di Down comporta un ritardo nello sviluppo fisico e mentale. Il quoziente intellettivo ha ampia variabilità intra-sindromica: raramente è normale e generalmente è compreso tra 50 e 80.
- Linguaggio e Apprendimento: Il linguaggio è la funzione strettamente legata allo sviluppo cognitivo che appare più compromessa rispetto all'organizzazione delle altre abilità superiori e al livello intellettivo. Tuttavia, grazie a una diagnosi precoce e a programmi sempre più su misura, sia educativi che riabilitativi, molti bambini acquisiscono buone competenze linguistiche e mostrano continui progressi nell'apprendimento.
- Comportamento: I neonati con sindrome di Down tendono a essere calmi e passivi e piangono meno del normale. Nel corso dell'infanzia si riscontra spesso un comportamento che indica disturbo da deficit di attenzione/iperattività, scarsa persistenza e distraibilità. I bambini affetti dalla sindrome di Down presentano anche un rischio più elevato di comportamenti di tipo autistico, soprattutto quelli con grave deficit intellettivo, e di sviluppare depressione e ansia in età adulta.
- Carattere e Socialità: Le caratteristiche caratteriali e comportamentali sono variabili, potendo cambiare in relazione all'età e allo stato di salute generale. Il carattere affettuoso e la socialità possono alternarsi a momenti di rigidità e ostinazione (soprattutto durante la crescita), rabbia e scatti d'ira.
L'ipotesi iniziale di Lejeune, negli anni '60, era che la presenza del cromosoma sovrannumerario comportasse un problema nel metabolismo cellulare con il risultato finale di intossicare i neuroni, causando così la disabilità intellettiva. Un recente articolo pubblicato su Scientific Reports, a opera del Professor Pierluigi Strippoli dell’Università di Bologna, ha individuato per la prima volta nel sangue e nelle urine dei bambini con sindrome di Down un profilo metabolico caratteristico, con sostanze rilevate in concentrazioni anomale proporzionali al modello previsto dai genetisti in presenza di un cromosoma in più.
È fondamentale intervenire tempestivamente con la fisioterapia, la terapia psicomotoria e la logopedia, che comprenda strumenti alternativi di comunicazione non verbale, in particolare la lingua dei segni e la comunicazione per scambio di immagini, per stimolare la comunicazione precoce e favorire lo sviluppo delle abilità orali. Non esistono terapie farmacologiche che possano curare la sindrome, ma l'unica terapia che permette al bambino di svilupparsi in modo armonico e di acquisire abilità scolastiche e sociali è quella riabilitativa, da iniziare fin dai primi mesi di vita.
Lo sviluppo psicomotorio di un bambino con la sindrome di Down - Intervista alla dott.ssa Valentini
Complicanze Mediche e Gestione della Salute
La sindrome di Down si associa a un aumento del rischio di complicanze mediche che persistono per tutta la vita, rendendo essenziale un follow-up medico individualizzato.
Malformazioni Congenite
Circa la metà dei pazienti presenta malformazioni, spesso già visibili durante il secondo trimestre di gravidanza tramite ecografia, che possono associarsi ad anomalie morfologiche minori.
- Cardiopatie Congenite: Interessano 20-76% dei nati vivi con sindrome di Down (rispetto al 6-8% della popolazione generale). I più comuni sono il difetto del setto interventricolare e il difetto del setto atrioventricolare.
- Problemi Gastrointestinali: Circa il 6% dei bambini presenta problemi a carico dell'apparato gastroenterico, come atresia o stenosi duodenali, malattia di Hirschsprung, ano imperforato. Anche la celiachia è più comune in questi bambini.
Altre Comorbidità Frequenti
- Problemi uditivi e oculari: La maggior parte dei soggetti con la sindrome presenta perdita dell'udito e le infezioni dell’orecchio sono molto comuni. Circa il 60% delle persone con sindrome di Down presenta problemi oculari come cataratte, glaucoma e strabismo.
- Instabilità delle articolazioni del collo: Le articolazioni del collo possono essere instabili (instabilità atlanto-assiale), determinando una compressione del midollo spinale, che può portare a cambiamenti nell’andatura, nell’uso delle braccia e delle mani, nella funzione intestinale e vescicale o debolezza.
- Problemi tiroidei e metabolici: Molte persone con sindrome di Down sviluppano malattie della tiroide (come l’ipotiroidismo) e il diabete.
- Infezioni e leucemia: Presentano un rischio più elevato di sviluppare infezioni e leucemia.
- Apnea ostruttiva del sonno: Un rischio molto più elevato di sviluppare apnea ostruttiva del sonno.
Gestione e Follow-up Medico
Non esiste una cura per la sindrome di Down, tuttavia alcuni sintomi e problemi specifici possono essere trattati. L’assistenza dei pazienti con sindrome di Down deve includere un follow-up medico individualizzato per identificare e trattare tempestivamente le eventuali complicanze, secondo le linee-guida disponibili.
- Interventi chirurgici: I medici possono riparare chirurgicamente alcuni difetti cardiaci e gastrointestinali.
- Trattamento delle patologie associate: Altre malattie (come ipotiroidismo, celiachia, diabete e leucemia) vengono trattate secondo necessità.
- Screening regolari: Dopo la diagnosi, vengono eseguiti esami specifici a determinati intervalli, includendo ecografia del cuore, esami del sangue per la funzione tiroidea, esame della vista, test dell’udito. Le misurazioni di altezza, peso e circonferenza cranica vengono registrate a ogni bilancio di salute utilizzando una tabella della crescita specifica. È raccomandata la valutazione dell’apnea ostruttiva del sonno.
- Valutazione neurologica: I bambini con dolore al collo o ai nervi, debolezza o altri sintomi neurologici devono essere sottoposti a radiografia delle articolazioni del collo per rilevare eventuale instabilità.
- Screening per adulti: Adolescenti e adulti con sindrome di Down devono essere sottoposti a screening a determinati intervalli per il diabete, l'ipotiroidismo e la malattia di Alzheimer.
Aspettativa di Vita e Invecchiamento
La sindrome di Down ha una prognosi migliore rispetto ad altri disturbi causati da un cromosoma supplementare. L’aspettativa di vita nelle nazioni ad alto reddito è oggi mediamente superiore ai 55-60 anni, con variabilità legata alle comorbidità e al miglioramento dell’assistenza sanitaria, e alcuni soggetti superano gli 80 anni. Il processo di invecchiamento può essere accelerato, e sintomi di demenza, quali perdita di memoria, ulteriore peggioramento delle capacità intellettive e cambiamenti della personalità, possono svilupparsi in età più giovane. La maggior parte dei decessi è dovuta a malattie cardiache, infezioni e leucemie.

Screening e Diagnosi Prenatale
La diagnosi prenatale può essere di screening (stima del rischio) o diagnostica (conferma citogenetica o genomica). Lo screening e la diagnosi prenatale devono essere offerti a tutte le persone in gravidanza con informazioni chiare su benefici, limiti e alternative. Il consenso informato e la decisione condivisa sono pilastri fondamentali, così come la tutela dei dati genetici (categoria speciale ai sensi del GDPR). Non esistono sintomi specifici nella madre che possano indicare la presenza della sindrome di Down nel feto.
Test di Screening (Stima del Rischio)
I test di screening pre-natali consentono di calcolare il rischio che il nascituro presenti anomalie cromosomiche, ma danno solo risposte di tipo probabilistico.
Test Combinato del I Trimestre (Tra 11+0 e 13+6 settimane):
- Ecografia con misurazione della translucenza nucale (NT): Eseguita quando la CRL (lunghezza vertice-sacro) è compresa fra 45 e 84 mm. Valuta lo spessore dell’accumulo di liquido a livello della nuca fetale. L’aumento dell’NT e l’assenza/ipoplasia dell’osso nasale aumentano il sospetto di Trisomia 21 nel primo trimestre, ed è visibile nel 70-75% dei feti.
- Valutazione di altri marker ecografici: Osso nasale, flusso nel dotto venoso, rigurgito tricuspide ove previsto dai protocolli.
- Marcatori biochimici materni: Analisi del sangue per la PAPP-A (proteina A plasmatica associata alla gravidanza) e l'hCG libera (frazione libera della gonadotropina corionica).
- Performance: Il “test combinato” offre una buona performance di screening per trisomia 21 e altre aneuploidie, con valori riportati in letteratura attorno all’82-90% di rilevazione a falsi positivi del 3-5% in contesti controllati. Il BI-TEST, che integra questi valori con l'età materna, può individuare il 65% dei feti affetti con un 5% di falsi positivi.
- TRITEST: Può essere eseguito tra la 15ª e la 17ª settimana di gestazione.
Screening Non Invasivo su Sangue Materno (NIPT/cfDNA):
- Disponibilità: È disponibile dalla 10ª settimana di gestazione (o da circa 10-11 settimane).
- Funzionamento: Analizza il DNA fetale libero circolante (cfDNA) nel sangue materno attraverso un prelievo ematico. Il cfDNA riflette il genoma placentare.
- Accuratezza: Per la trisomia 21 la sensibilità e specificità sono molto elevate (circa >99% e >99% rispettivamente in numerosi studi). L’accuratezza sale quasi al 99%.
- Limiti: Il cfDNA resta uno screening: un risultato “ad alto rischio” non è una diagnosi e va confermato con test diagnostico invasivo prima di decisioni cliniche importanti. Inoltre, il valore predittivo positivo varia con il rischio pre-test (età materna, storia, esito di altri screening). Un risultato negativo non azzera il rischio.
- Fattori che possono influenzare il risultato: Fattori che riducono la fetal fraction (la percentuale di DNA fetale nel sangue materno) includono BMI elevato, prelievo troppo precoce, patologie autoimmuni e, in alcuni studi, terapia anticoagulante (eparina), che possono aumentare il tasso di risultati indeterminati ("no-call"). Mosaicismo placentare confinato, mosaicismo materno o raramente condizioni materne (p. es. neoplasie) possono spiegare risultati cfDNA discordanti con il cariotipo fetale.
- Costo e Accesso in Italia: In Italia non è gratuito, tranne in alcune regioni dove è stato inserito nei livelli essenziali di assistenza (LEA) tramite SSN (universalità, criteri di accesso, fasce di rischio o progetti pilota). In alternativa, è disponibile in intramoenia/privato.
- Analisi aggiuntive: Alcuni test cfDNA offrono l’analisi “genome-wide” di rare autosomal trisomies e microdelezioni.
Ecografia Morfologica (Tra 18 e 22 settimane):
- Raccomandazione: Raccomandata per tutte le gravidanze.
- Rilevazione: Può rilevare caratteristiche congenite legate alla sindrome di Down o malformazioni maggiori (es. cardiopatie). Il riscontro di soft marker isolati (p. es. cisti plesso coroideo, pielectasia) nel secondo trimestre ha valore aggiunto limitato se lo screening è negativo.
- Limite: Non conferma la sindrome senza un test combinato o un test del DNA fetale a precedere.
Diagnosi Confermativa (Test Invasivi)
Se i test di screening indicano un rischio elevato, la diagnosi può essere confermata con test invasivi che analizzano direttamente il cariotipo fetale.
- Villocentesi: Si esegue in genere tra 10 e 13 settimane e campiona i villi coriali placentari.
- Amniocentesi: Si esegue dal secondo trimestre (≥15 settimane) e campiona liquido amniotico. Consiste nel prelievo di circa 15-30 ml di liquido amniotico, attraverso un ago introdotto nell’addome materno sotto monitoraggio ecografico.
- Cordonocentesi: Prelievo di sangue fetale che si effettua dopo la 18ª settimana di gestazione.
Entrambe le procedure (villocentesi e amniocentesi) consentono la diagnosi citogenetica o genomica del feto. Nelle coorti moderne con operatori esperti il rischio aggiuntivo di perdita fetale è basso (ordine di pochi per mille). In presenza di malformazioni fetali all’ecografia, il microarray è raccomandato perché aumenta il tasso diagnostico rispetto al cariotipo standard. Indipendentemente dall’età, a tutte le gestanti è consigliato l’esame per la sindrome di Down prima della 20ª settimana di gestazione.

Diagnosi Post-Natale e Supporto alla Persona con Sindrome di Down
Dopo la nascita, la diagnosi è suggerita dall’aspetto fisico del bambino ed è confermata dall’individuazione di una terza copia del cromosoma 21, di solito mediante l’analisi di un campione di sangue per il cariotipo.
Diagnosi e Cura Post-Natale
La diagnosi post-natale nei neonati si basa sull’osservazione delle caratteristiche cliniche, di sintomi legati a patologie congenite già diagnosticate in gravidanza o di sintomi nuovi. Viene eseguito anche il test della conferma del cariotipo per il numero dei cromosomi e viene valutata la possibile coesistenza di altre patologie genetiche o congenite, non diagnosticabili nel periodo prenatale. Subito dopo la diagnosi, i medici eseguono altri esami per individuare le anomalie associate alla sindrome di Down. Il trattamento delle anomalie rilevate può spesso prevenire il peggioramento delle condizioni di salute.
La presa in cura multidisciplinare è necessaria. Il follow-up medico individualizzato è fondamentale per identificare e trattare tempestivamente le eventuali complicanze, secondo le linee-guida disponibili. Può rendersi necessario il mantenimento del supporto anche nell’età adulta, compresa la rieducazione.
Prevenzione e Supporto
- Prevenzione primaria (preconcezionale): Non è possibile attuare una prevenzione primaria per la sindrome di Down, ma è fondamentale l'informazione e il counseling genetico nelle coppie con storia familiare rilevante, ad esempio, per valutare la possibilità di rischio che una coppia ha nel procreare.
- Inclusione Sociale e Autonomia: Nel percorso di crescita, l’aspetto sociale e pedagogico riveste un ruolo essenziale. Convivere con la sindrome di Down oggi significa affrontare delle sfide ma anche scoprire risorse preziose. I genitori e i familiari cercano di favorire l’inclusione sociale e l’autonomia dei propri figli anche attraverso opportunità di inserimento lavorativo, un ambito in cui la società deve ancora compiere passi avanti, superando pregiudizi e mancanza di accoglienza. Il paziente deve essere coinvolto quanto prima nel processo decisionale attraverso l'autodeterminazione.
- Programmi di Riabilitazione e Scolarizzazione: Si raccomanda di predisporre un programma adatto, che preveda la rieducazione, la scolarizzazione e le competenze sociali, per favorire la migliore integrazione sociale possibile (ad es. oltre la metà dei pazienti è in grado di leggere e scrivere, anche parzialmente). Le valutazioni neuropsichiatriche sono necessarie per individuare le difficoltà e le abilità individuali e, di conseguenza, per suggerire strategie cognitive mirate.
- Servizio Sanitario Nazionale (SSN): Le prestazioni per la tutela della maternità rientrano nei Livelli Essenziali di Assistenza (LEA) e sono in esenzione ticket secondo calendari e condizioni definite (codici “M” indicati sulle ricette dematerializzate). L’offerta del NIPT tramite SSN varia per Regione.

Rischio di Ricorrenza in Future Gravidanze
Comprendere il rischio di ricorrenza è fondamentale per le famiglie che hanno già avuto un bambino con sindrome di Down. Il rischio varia a seconda del tipo genetico della trisomia 21.
Trisomia 21 Libera da Non-Disgiunzione
Dopo una gravidanza con trisomia 21 “libera”, il rischio in una successiva gravidanza è circa dell'1% oltre al rischio legato all’età materna (o il maggiore fra i due). Per i genitori e i familiari di un bambino con trisomia 21 libera, il rischio di ricorrenza non s’incrementa in modo significativo rispetto a quello della popolazione generale, rimanendo circa l'1% fino ai 40 anni di età e, successivamente, in relazione all'età materna.
Trisomia 21 da Traslocazione Robertsoniana
Se la trisomia 21 è dovuta a traslocazione Robertsoniana, è indicato il cariotipo dei genitori.
- Genitori con cariotipo normale: Se i genitori non sono portatori di una traslocazione bilanciata, il rischio residuo è in genere di circa il 2-3%.
- Un genitore portatore di traslocazione bilanciata: In questo caso, il rischio di ricorrenza sale considerevolmente e dipende dal sesso del portatore e dal cromosoma coinvolto. Se la madre è portatrice, il rischio è del 10-15%; se il padre è portatore, il rischio è di circa l'1-3%. Questa condizione non è collegata all'età. È possibile che anche i fratelli e le sorelle di un bambino con sindrome di Down da traslocazione siano portatori della traslocazione bilanciata, ed essere quindi soggetti ad un rischio aumentato per le loro gravidanze.
- Traslocazione 21;21: Una traslocazione specifica 21;21 implica un rischio molto elevato di prole affetta, potenzialmente quasi il 100% di nati vivi affetti o aborti spontanei ricorrenti, a seconda della segregazione meiotica. Il counseling genetico è cruciale in questi casi.
Un genetista ha il compito di valutare la possibilità di rischio che ha una coppia nel procreare, un servizio particolarmente importante per le malattie genetiche come la sindrome di Down, dove la prevenzione primaria è impraticabile ma la diagnosi prenatale permette di diagnosticare il cariotipo fetale in gravidanze ad alto rischio.
tags: #sindrome #down #trisomia #libera #rischio #ricorrenza