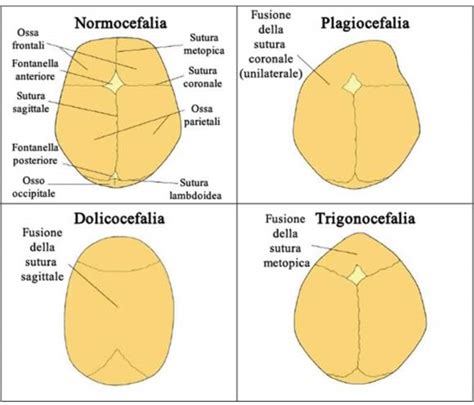
La Sindrome di Apert è una rara malattia genetica appartenente al gruppo delle acrocefalosindattilie (tipo I). Si manifesta fin dalla nascita ed è caratterizzata da una triade clinica distintiva: la chiusura precoce delle suture craniche, gravi malformazioni del massiccio facciale e la fusione delle dita delle mani e dei piedi. Dal punto di vista fisiopatologico, la sindrome impedisce al cranio di crescere normalmente, costringendo il cervello in espansione a esercitare pressione sulle ossa craniche e a svilupparsi in direzioni non fisiologiche. Questo porta a una forma del capo alterata, spesso descritta come cranio a torre o cranio corto e largo.
Eziologia e basi genetiche
La Sindrome di Apert è causata da mutazioni specifiche nel gene FGFR2 (Fibroblast Growth Factor Receptor 2), situato sul cromosoma 10 (10q25.3-10q26). Questo gene è responsabile della produzione di una proteina che segnala alle cellule immature di trasformarsi in cellule ossee durante lo sviluppo embrionale. Le mutazioni responsabili della sindrome di Apert si localizzano su un gene noto come FGFR2, che è responsabile delle istruzioni per la sintesi di una proteina chiamata “recettore 2 del fattore di crescita dei fibroblasti”.
Nella stragrande maggioranza dei casi (circa il 95-98%), la sindrome si verifica in modo sporadico, ovvero come una mutazione "de novo" che avviene durante la formazione dei gameti o nelle prime fasi dello sviluppo embrionale, senza che i genitori presentino la malattia. Un fattore di rischio statisticamente rilevante identificato dagli studi epidemiologici è l'età paterna avanzata. È stato osservato che il rischio di mutazioni spontanee nel gene FGFR2 aumenta con l'aumentare dell'età del padre, probabilmente a causa di errori cumulativi nella replicazione del DNA durante la produzione degli spermatozoi. La sindrome di Apert ha una trasmissione autosomica dominante a penetranza completa. Per i genitori non affetti dei bambini che presentano la sindrome, il rischio di ricorrenza è basso, mentre una persona affetta ha una probabilità del 50% di trasmettere la malattia ai propri figli.
Quadro clinico e manifestazioni fenotipiche
I pazienti in genere presentano deficit strutturali e funzionali diffusi associati a deformità del cranio e degli arti. La craniosinostosi può portare all'acrobrachicefalia o alla turribrachicefalia con chiusura ritardata delle fontanelle e un possibile impatto sulla crescita cerebrale e lo sviluppo neurologico. È stata descritta anche macrocefalia. Deformità cranica: la chiusura precoce delle suture craniche, nota come craniostenosi, è una delle caratteristiche distintive della sindrome. Questo fenomeno porta a un appiattimento della fronte e della parte posteriore del cranio (brachicefalia), accompagnato da una crescita verticale eccessiva del cranio (acro/turricefalia).
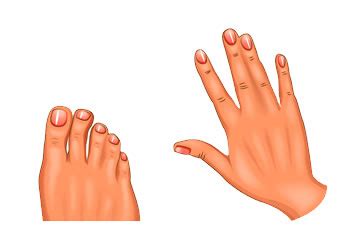
Le malformazioni degli arti consistono essenzialmente nella sindattilia dei tessuti molli e delle ossa delle dita di mani e piedi (che interessa un numero variabile di dita), in un occasionale accorciamento rizomelico, nell'anchilosi del gomito, con deficit funzionale e limitazione della mobilità. L'altra caratteristica tipica della condizione è la sindattilia, ovvero la presenza di dita (di mani e/o piedi) fuse insieme; la gravità della fusione è variabile, sebbene le mani tendano ad essere più gravemente colpite rispetto ai piedi. Nei casi tipici, si osserva la fusione cutanea e ossea del secondo, terzo e quarto dito, creando quella che viene definita "mano a guanto" o "mano a cucchiaio".
Le caratteristiche facciali comprendono l'ipoplasia mediofacciale, che è in genere moderata-grave e si associa a ipoplasia della mascella, orbite poco profonde, strabismo, ipertelorismo, rime palpebrali oblique verso il basso, proptosi, radice del naso depressa e deviazione del setto nasale. I segni dentali comprendono il ritardo dell'eruzione, l'occlusione, l'affollamento, un significativo gonfiore gengivale, l'agenesia di alcuni denti e l'elevato rischio di carie. È presente un morso crociato posteriore monolaterale e bilaterale.
Diagnosi prenatale e tecniche molecolari
Grazie alle moderne tecnologie di imaging, è possibile sospettare la sindrome durante la gravidanza. L'ecografia morfologica del secondo trimestre può rilevare anomalie nella forma del cranio (come il "cranio a trifoglio"), l'ipertelorismo o la sindattilia delle mani. L'approccio tradizionale nella diagnosi prenatale di anomalie cromosomiche comporta la messa in coltura di cellule fetali ricavate da prelievi di liquido amniotico e la determinazione del cariotipo tramite l'analisi al microscopio dei cromosomi in metafase.
Oggi si utilizza una tecnica di diagnostica molecolare che permette diagnosi rapida di alcune aneuploidie fetali, conosciuta come amplificazione enzimatica in vitro del DNA fluorescente o QF-PCR (Quantitative Fluorescence PCR). Tale tecnica si affianca alle analisi citogenetiche tradizionali, differenziandosi per una maggiore rapidità (risultato disponibile entro 24h-48h dal prelievo) e per la necessità di una minore quantità di materiale biologico. Tuttavia, il cariotipo tradizionale non garantisce che il feto sia esente da malattie genetiche o alterazioni cromosomiche di piccole dimensioni.
Rispetto all'esame citogenetico tradizionale, l'analisi molecolare (come l'array-CGH) ha una risoluzione molto più elevata. Ciò consente di identificare alcune patologie derivanti da alterazioni cromosomiche submicroscopiche (microdelezioni e microduplicazioni), aumentando sensibilmente l'accuratezza dell'esame. L'analisi array-CGH rappresenta una tecnica ideale di approfondimento diagnostico di II livello, eseguita per integrare l'analisi citogenetica prenatale al fine di definire più accuratamente eventuali anomalie cromosomiche.
La reazione a catena della polimerasi (PCR)
Gestione clinica e approccio multidisciplinare
Il trattamento della Sindrome di Apert è estremamente complesso e richiede un team multidisciplinare composto da neurochirurghi, chirurghi plastici, maxillo-facciali, otorinolaringoiatri, odontoiatri, logopedisti e psicologi. Secondo il Dott. Mario Igor Rossello, la sindrome di Apert è una patologia complessa che può richiedere interventi chirurgici avanzati e una diagnosi precoce per salvaguardare la qualità di vita del paziente.
L'obiettivo primario è rilasciare le suture fuse per permettere al cervello di crescere e ridurre la pressione intracranica. Questo intervento, chiamato espansione del vault cranico, viene solitamente eseguito tra i 6 e i 12 mesi di vita, ma può essere necessario anticipare i tempi nel caso sia presente un'ipertensione endocranica. Per correggere l'ipoplasia della parte media del volto e migliorare la respirazione, si ricorre spesso all'osteotomia di tipo Le Fort III. La separazione delle dita (rilascio della sindattilia) viene pianificata in più fasi, solitamente iniziando entro il primo o secondo anno di vita.
Le complicazioni correlate più comuni sono l'otite media cronica, la sordità e l'aumento della pressione oculare, che può causare cecità. Inoltre, a causa della conformazione del massiccio facciale e delle vie aeree ristrette, molti pazienti soffrono di apnee ostruttive del sonno. Sebbene molti individui con Sindrome di Apert abbiano un'intelligenza normale, esiste un rischio aumentato di disabilità intellettiva o difficoltà di apprendimento, spesso correlate alla pressione intracranica elevata o a malformazioni cerebrali associate come l'idrocefalo.
Prognosi e follow-up a lungo termine
La prognosi per i bambini nati con la Sindrome di Apert è migliorata drasticamente negli ultimi decenni. Con un intervento chirurgico precoce e un monitoraggio costante, la maggior parte dei pazienti può condurre una vita lunga e produttiva. Il decorso cognitivo è variabile: sebbene esista una maggiore incidenza di ritardi nello sviluppo, molti individui raggiungono livelli di istruzione superiori e l'indipendenza lavorativa.
Il successo scolastico e sociale dipende spesso dalla precocità degli interventi neurochirurgici e dalla qualità del supporto educativo ricevuto. Dal punto di vista sociale, l'adolescenza può essere un periodo critico a causa delle differenze fisiche. Poiché la Sindrome di Apert è causata da una mutazione genetica spontanea nella maggior parte dei casi, non esiste una prevenzione primaria efficace. L'unica forma di prevenzione è quella secondaria, basata sulla consulenza genetica. Le famiglie che hanno già un bambino con Sindrome di Apert possono consultare un genetista per valutare il rischio di ricorrenza, che per i genitori sani è estremamente basso, ma non nullo a causa del possibile mosaicismo germinale.
Il trattamento efficace può migliorare l'estetica e la prestazione funzionale (respirazione, masticazione, salute orale e oculare). Nei pazienti con sindattilia tipo "guanto a manopola", la separazione chirurgica delle dita permette in genere un lieve miglioramento funzionale. La prognosi deve essere valutata con cautela, poiché molti pazienti presentano complicazioni potenzialmente letali, che comprendono la compromissione delle vie aeree e del sistema nervoso centrale. Tuttavia, altri pazienti possono avere una buona prognosi in presenza di una presa in carico medica e chirurgica adeguata, anche se le limitazioni cognitive rimangono sempre molto frequenti. Il follow-up deve essere garantito per tutto l'arco della vita del paziente, assicurando che ogni aspetto, dallo sviluppo neurologico alla salute delle vie aeree, sia monitorato da specialisti esperti.
tags: #sindrome #di #apert #amniocentesi