L'ossigeno è un elemento vitale per la maggior parte degli organismi viventi, e il suo efficiente trasporto e immagazzinamento sono processi fondamentali regolati da proteine specializzate. Tra queste, l'emoglobina (Hb) e la mioglobina (Mb) svolgono ruoli centrali, sebbene con meccanismi e contesti funzionali distinti. Comprendere le loro differenze strutturali e funzionali, in particolare tra le varianti fetali e adulte dell'emoglobina, è essenziale per apprezzare l'adattamento biologico a diverse esigenze fisiologiche, dalla vita intrauterina alle sfide dell'altitudine o alle condizioni patologiche.
Mioglobina e Emoglobina: Un Confronto Strutturale e Funzionale
Per poter parlare in modo comprensibile dell'emoglobina (Hb), è utile occuparsi prima della mioglobina (Mb) che è molto simile all'emoglobina ma è molto più semplice. Tra emoglobina e mioglobina ci sono stringenti relazioni di parentela: entrambe sono proteine coniugate ed il loro gruppo prostetico (parte non proteica) è il gruppo eme.
La mioglobina è una proteina globulare costituita da una singola catena di circa centocinquanta amminoacidi (dipende dall'organismo) ed il suo peso molecolare è di circa 18 Kd.
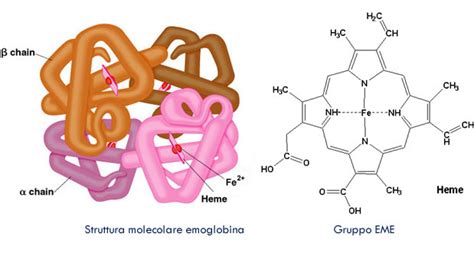
L'emoglobina (indicata con le sigla Hb o hgb, dall'inglese hemoglobin o haemoglobin) è una proteina globulare con struttura quaternaria formata da quattro subunità ed è solubile e di colore rosso (cromoproteina). L’emoglobina (abbreviata Hb) è una proteina appartenente alla classe delle globine e come la maggior parte delle proteine di questa classe, svolge funzione di immagazzinatore di ossigeno, ma la sua funzione principale è il trasporto di ossigeno dai polmoni ai tessuti periferici. L'emoglobina è un tetramero, cioè è costituita da quattro catene polipeptidiche ciascuna dotata di un gruppo eme ed identiche a due a due. Le catene del tetramero fisiologicamente sono a due a due uguali: due appartenenti alla classe α (in giallo nella figura) e due alla classe β (in rosso). La struttura quaternaria è un tetramero costituito da due catene α di 141 residui ciascuna e di due catene β di 146 residui ciascuna. Del gruppo α fanno parte le catene α1, α2 e ζ mentre del gruppo β fanno parte le catene β, Aγ, Gγ (queste due molto simili per struttura ed espressione), δ, e infine ε. I monomeri hanno una struttura secondaria quasi totalmente identica nonostante gli amminoacidi in comune siano intorno al 20%. Sono tutte formate da otto α eliche (indicate con le lettere da A a H) intervallate da brevi segmenti di congiunzione indicate con la coppia di lettere delle eliche che la precedono e la seguono (es. ansa BC). Ognuna delle quattro catene trasporta un gruppo eme. L’emoglobina è in grado di legare e trasportare anche protoni (H+) o molecole di CO2.
L'emoglobina si lega all'ossigeno in modo più debole rispetto alla mioglobina e lo rilascia più facilmente, per questo motivo non ha un andamento acceso-spento ma presenta delle condizioni intermedie e la sua rappresentazione cinetica è una curva sigmoide (blu nell’immagine). L'emoglobina, infatti, ha un comportamento più elastico: lega l'ossigeno ad alte pressioni e lo rilascia quando la pressione diminuisce. La mioglobina è efficiente nel legame ma non nel trasporto mentre l’emoglobina è efficiente in entrambe le cose. Una spiegazione della presenza di più catene diverse nell'emoglobina è la seguente: nel corso del processo evolutivo degli organismi, anche l'emoglobina si è evoluta specializzandosi nel trasporto di ossigeno da zone che ne sono ricche a zone carenti.
Il Gruppo Eme: Il Cuore del Legame con l'Ossigeno
Che cos’è il gruppo eme? Ogni monomero ospita in una tasca idrofobica il gruppo prostetico (il gruppo eme), vero cuore della macromolecola, legato covalentemente con un'istidina responsabile di importanti funzioni che verranno esaminate in seguito. Ognuna delle sue quattro catene polipeptidiche è legata covalentemente a un gruppo prostetico detto eme, costituito da una molecola di protoporfirina IX (che rappresenta la componente organica) coordinante uno ione ferro Fe2+ (rappresentante la componente inorganica) che sporge leggermente dal piano della molecola.
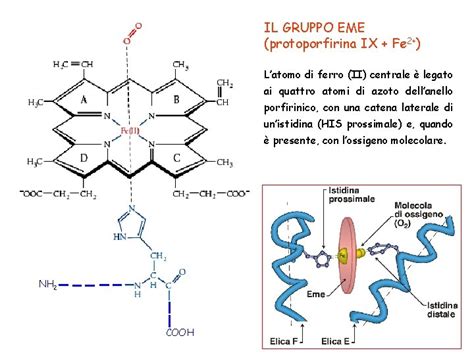
Il gruppo eme è costituito da una molecola organica complessa, la protoporfirina, che coordina uno ione ferroso (Fe2+). Questo metallo è in grado di formare sei legami di coordinazione: quattro sono utilizzati per legare l’azoto degli anelli pirrolici e altri due perpendicolari alla protoporfirina che legano da un lato un residuo di istidina della proteina e dall’altro lega in modo reversibile una molecola di ossigeno. Il legame tra protoporfirina e ferro è un legame tipico dei composti detti di coordinazione che sono composti chimici in cui un atomo (o ione) centrale, forma dei legami con altre specie chimiche in numero superiore al suo numero di ossidazione (carica elettrica). Il numero di coordinazione (numero di legami di coordinazione) del ferro è sei: ci possono essere sei molecole attorno al ferro che mettono in condivisione gli elettroni di legame. Quando il ferro è sotto forma di ione libero, i suoi orbitali di tipo d hanno tutti la stessa energia; nella mioglobina, lo ione ferro è legato alla protoporfirina e all'istidina: tali specie perturbano magneticamente gli orbitali d del ferro; l'entità della perturbazione sarà diversa per i vari orbitali d a seconda della loro orientazione spaziale e di quella delle specie perturbanti. Interazioni ioniche e idrofobiche e interazioni π-π con un residuo di fenilalanina contribuiscono a mantenere l'eme in posizione.
La zona in cui si lega l’ossigeno è una zona apolare che si trova in una sacca della struttura terziaria. Spazialmente, vicino all’ossigeno legato al gruppo Fe2+, troviamo un istidina detta istidina distale che protegge il gruppo Eme impedendo che si formino ponti ossigeno Eme-O-Eme. Inoltre, l’istidina distale abbassa l’affinità del gruppo Eme per sostanze come il cianuro (CN-) e il monossido di carbonio (CO) che vi si legano in modo quasi irreversibile causando soffocamento.
Il Meccanismo di Legame dell'Ossigeno e i Suoi Modulatori Allosterici
Durante l’atto inspiratorio riempiamo i polmoni di ossigeno, questo negli alveoli passa nel sangue e nello specifico si lega all’emoglobina dentro gli eritrociti. L’ossigeno è poco solubile in acqua, è una molecola idrofoba che nel sangue può avere una concentrazione di 7,15 mg/L mentre l’emoglobina ne porta 357 mg/L. L’emoglobina lo trasporta a tutti i tessuti poiché l’ossigeno è necessario a tutte le cellule per compiere la respirazione cellulare e produrre energia. Le reazioni di scambio gassoso sono rese possibili dalla presenza di ioni ferro in ogni gruppo. Questo cofattore metallico nella sua forma Fe2+ lega l'ossigeno durante il passaggio del sangue nei polmoni e lo cede successivamente ai tessuti nella circolazione periferica.
Conformazioni Tesa (T) e Rilassata (R)
L’emoglobina può esistere in due stati conformazionali differenti: T (tesa) e R (rilassata). Lo stato R ha una maggior affinità per l’ossigeno e quindi sarà la forma prevalente dell’ossiemoglobina. Lo stato T è la conformazione della deossiemoglobina. Nella forma T il gruppo eme tende ad assumere una forma a cupola dove l’atomo di Fe viene ad essere attratto dall’istidina prossimale; nella forma R il gruppo eme e l’atomo di Fe sono sullo stesso piano. L'emoglobina presenta due strutture limite, la prima prevalentemente rappresentata dalla deossiemoglobina, definita come "struttura tesa" (T), la seconda relativa, invece, alla ossiemoglobina, chiamata questa volta "struttura rilassata" (R). Queste due "forme" sono caratterizzate da una differente affinità per l'ossigeno, infatti la struttura T presenta un'affinità minore rispetto alla struttura R.
La possibilità di modificare il proprio stato di transizione determina una curva di legame all’ossigeno sigmoide. Questo determina una bassa affinità all’ossigeno a basse pressioni parziali e un’alta affinità ad alte pressioni parziali di ossigeno (curva verde). La struttura quaternaria della deossiemoglobina prende il nome di forma T (tesa) mentre quella della ossiemoglobina viene chiamata forma R (rilasciata); nello stato teso vi sono una serie di interazioni elettrostatiche piuttosto forti tra amminoacidi acidi e amminoacidi basici che portano ad una struttura rigida della deossiemoglobina (ecco il perché del "forma tesa"), mentre quando si lega l'ossigeno, l'entità di queste interazioni diminuisce (ecco il perché del "forma rilasciata"). Quando il gruppo prostetico, al legame con l'ossigeno, subisce un cambiamento, si appiattisce. L'emoglobina è inoltre una proteina allosterica.
La Cooperatività del Legame
Quando si fa riferimento alla cooperatività dell’emoglobina si intende il fatto che proteina e ossigeno cooperino per far sì che essa si carichi o scarichi di ossigeno: ad ogni ossigeno che lega aumenta la probabilità che l’emoglobina diventi in forma R; il fatto che sia in forma R aumenta la probabilità di legare ossigeno. Questo si verifica a livello alveolare permettendo la cattura dell'ossigeno da parte dell'emoglobina, mentre il contrario succede a livello periferico, dove l'emoglobina passa dallo stato R allo stato T rilasciando ossigeno ai tessuti che possono così assumerlo per utilizzarlo attraverso le ossidazioni. Quando una proteina è funzionalmente attiva, essa può mutare un po' la sua forma; ad esempio la mioglobina ossigenata ha una forma diversa dalla mioglobina non ossigenata e questa mutazione non influisce su quelle vicine. Il discorso è diverso nel caso di proteine associate come l'emoglobina: quando una catena si ossigena è indotta a cambiare la sua forma ma tale modificazione è tridimensionale perciò ne risentono anche le altre catene del tetramero. Il fatto che le catene siano tra loro associate, induce a pensare che la modifica di una si ripercuota sulle altre vicine anche se in misura diversa; quando una catena si ossigena, le altre catene del tetramero assumono un "atteggiamento meno ostile" nei riguardi dell'ossigeno: la difficoltà con cui una catena si ossigena diminuisce man mano che le catene ad essa vicine si ossigenano a loro volta.
La saturazione indica il rapporto percentuale tra il numero medio di molecole di ossigeno (O2) realmente legate alle molecole di emoglobina, e il massimo numero di molecole di ossigeno che potrebbero essere legate alle stesse molecole di emoglobina. Ogni molecola di emoglobina è in grado di legarsi, al massimo, con quattro molecole di O2 e quando ciò accade si dice sia satura. Sebbene esista una piccola percentuale di ossigeno non legata all'emoglobina (pari a circa 0,3 mL/100 mL di sangue, lo 0,3%), il resto dell'ossigeno è legato e trasportato dall'emoglobina.
Il Ruolo del 2,3-Bisfosfoglicerato (BPG)
Il 2,3-bisfosfoglicerato (2,3-BPG, isomero dell’1,3 BPG che è un intermedio della glicolisi) è un composto anionico presente negli eritrociti approssimativamente alla stessa concentrazione dell’emoglobina. Questo modulatore allosterico eterotropico si lega in un punto diverso dal sito attivo ed è una molecola diversa dal comune substrato della proteina. Questa molecola si lega all’emoglobina in modo allosterico: si lega, cioè, in un sito diverso da quello principale dell’ossigeno (gruppo Eme) limitando però l’attitudine della proteina a legarsi a quest’ultimo. In altre parole, diminuisce l’affinità per l’ossigeno stabilizzando la forma T (deossi-emoglobina) e lo fa legandosi in uno spazio “vuoto” tra le quattro subunità di emoglobina che è più ampio in forma T e meno ampio nella forma R. A pH fisiologico, il 2,3 bisfosfoglicerato è deprotonato ed ha su di sé cinque cariche negative; si va ad incuneare tra le due catene beta dell'emoglobina perché tali catene presentano un'elevata concentrazione di cariche positive.
La saturazione dell'emoglobina per l'ossigeno è influenzata dalla pressione parziale dell'ossigeno che con legame cooperativo aumenta l'affinità dell'emoglobina verso l'ossigeno in risposta a un aumento della quantità di ossigeno. Il 2,3-bisfosfoglicerato, presente nei globuli rossi, riduce l'affinità dell'emoglobina per l'ossigeno perché stabilizza la sua forma deossigenata. La sua quantità presente nel globulo rosso è regolata dalla pressione parziale dell'ossigeno nell'ambiente per un ottimale rilascio ai tessuti.
Il 2,3-bisfosfoglicerato è utile anche in condizioni di ipossia o di basse pressioni parziali di ossigeno nei polmoni (come ad esempio in alta quota). Per cui se una persona che vive a livello del mare si sposta in montagna, dove la pressione dell'ossigeno è minore, ha nei primi giorni una carenza di BPG e quindi una ridotta capacità a fare lavoro, perché l'emoglobina ha un'elevata affinità per l'ossigeno e non ne rilascia abbastanza nei tessuti. Man mano che la concentrazione di BPG aumenta nei suoi eritrociti diminuendo l'affinità per l'ossigeno, può fare più lavoro e si dice che acquista il "passo del montanaro".
L'Effetto Bohr e l'Anidride Carbonica
Come prodotto di scarto della respirazione cellulare si ottiene anidride carbonica (CO2) che entra nel globulo rosso e qui viene trasformato in ione idrogenocarbonato (HCO3-) dall’enzima anidrasi carbonica (in grado di trasformare 106 molecole di CO2 al secondo) secondo la reazione: CO2 + H2O ⇌ H++ HCO3-. Questa reazione libera ioni idronio (H+) che abbassano il pH (lo rendono più acido). Questo è molto importante perché il pH acido rende l’emoglobina meno affine all’ossigeno. Perciò, giungendo ai tessuti periferici dove c’è un'alta concentrazione di anidride carbonica che si traduce in un abbassamento del pH all’interno dell’eritrocita, l’emoglobina è spinta al rilascio di ossigeno a quegli stessi tessuti. Al contrario quando la CO2 raggiunge i polmoni e viene rilasciata, il pH sale e l’affinità dell’emoglobina per l’ossigeno cresce. Questo è noto come effetto Bohr.
Inoltre, in assenza di ossigeno, la carica dell'istidina (vedi struttura) viene stabilizzata dalla carica opposta dell'acido aspartico mentre, in presenza di ossigeno, c'è la tendenza da parte della proteina, a perdere un protone; tutto ciò comporta che l'emoglobina ossigenata sia un acido più forte dell'emoglobina deossigenata. Ogni emoglobina rilascia 0,7 protoni per mole di ossigeno (O2) entrante. L'efficacia dell'emoglobina è influenzata anche dalla maggiore presenza di CO2 a livello dei tessuti che, grazie al suo comportamento acido in acqua, rilascia protoni che si legano all'emoglobina favorendo la formazione di ponti salini caratteristici della conformazione T; si ha quindi il rilascio di ossigeno. Inoltre, la CO2 che non si è ancora dissociata si lega ai gruppi amminici delle catene β dell'emoglobina formando così la carbodiossiemoglobina e quindi si ha il rilascio di ossigeno da parte della stessa.
Effetti Aggiuntivi: Temperatura e Trasporto Ionico
Vediamo l'effetto della temperatura. Altre influenze sulla saturazione dell'emoglobina per l'ossigeno sono: la temperatura.Un altro fenomeno che si verifica quando un eritrocita raggiunge un tessuto è il seguente: per gradiente, l'HCO3- (derivato dell'anidride carbonica) esce dall'eritrocita e, per bilanciare l'uscita di una carica negativa, si ha l'ingresso di cloruri che determina un aumento della pressione osmotica: per bilanciare questa variazione si verifica anche l'ingresso di acqua che causa un rigonfiamento dell'eritrocita (effetto HAMBURGER). Il fenomeno opposto si verifica quando un eritrocita raggiunge gli alveoli polmonari: si ha uno sgonfiamento degli eritrociti (effetto HALDANE).
Emoglobina Anomala e Tossicità
Fisiologicamente o per azione di determinate sostanze ossidanti (acqua ossigenata, permanganato di potassio, nitrito, ecc.) o di certi farmaci o di alcune sostanze contenute nelle fave (vedi favismo), lo ione Fe2+ (ione ferroso) si trasforma in ione Fe3+ (ione ferrico) e l'emoglobina si trasforma in metaemoglobina (MetHb), incapace di legare l'ossigeno. Nel sangue è normale una presenza del 2% di MetHb; se questa percentuale sale, la respirazione viene compromessa. Un altro pericolo per la respirazione è il monossido di carbonio (CO): l'emoglobina ha un'affinità circa 250 volte superiore per questo gas che per l'ossigeno, si lega quindi rapidamente al CO e il legame formato è estremamente difficile da scindere (quasi irreversibile normalmente). Se nell'aria è presente una percentuale di CO pari a 1/250 quella dell'ossigeno (circa quattro parti per mille di ossigeno), la metà dell'emoglobina si combinerà con l'ossido di carbonio, dando luogo a carbossiemoglobina (HbCO), incapace di legare ossigeno.
Mioglobina: funzioni e differenze rispetto all'Emoglobina 🩸
Emoglobina Fetale (HbF) vs. Emoglobina Adulta (HbA): L'Adattamento Cruciale per la Vita Pre-Natale
Nello sviluppo fetale l’emoglobina del feto (HbF) svolge un ruolo chiave in quanto è richiesto un elevato numero di molecole di ossigeno per la crescita del feto. L’emoglobina fetale (HbF) è la prima emoglobina prodotta durante la vita fetale. Questa è nota con il nome di emoglobina fetale, rappresenta l’1-2% dell’Hb dell’adulto ed è costituita da due catene alfa e due catene gamma (α2γ2). Normalmente la produzione di HbF diminuisce progressivamente alla nascita per poi stabilizzarsi su valori molto bassi intorno a 1-2 anni.
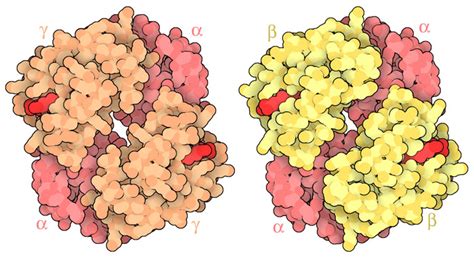
La differenza principale consiste nella predominanza dell'emoglobina fetale (HbF) con un'affinità, in vivo, molto elevata per l'O2. Questa differenza porta, in condizioni fisiologiche, a un passaggio favorevole dell'O2 dall'HbA (emoglobina dell'adulto) della madre all'HbF del feto. Questo processo non è affatto semplice. Il fatto non è stato chiarito fino alla scoperta che un'altra molecola, il 2,3-bisfosfoglicerato (BPG), è presente in condizioni fisiologiche e si lega alle deossiemoglobine. Il 2,3-BPG svolge anche un importante ruolo nell'approvvigionamento di ossigeno al feto. L'emoglobina fetale ha un'affinità maggiore per l'ossigeno rispetto all'emoglobina adulta. Ciò è essenziale per il trasferimento di ossigeno dalla madre al feto.
L'HbF lega più debolmente il BPG della HbA, dal momento che le due subunità γ dell'emoglobina fetale contengono un numero inferiore di amminoacidi capaci di carica positiva che possano interagire con le cariche negative del 2,3-BPG. Poiché il 2,3-BPG diminuisce l'affinità per l'O2 delle emoglobine il risultato nella HbF è un aumento dell'affinità per l'O2 della stessa. Questo consente al feto di "strappare" ossigeno dall'emoglobina adulta della madre. Di conseguenza, l'emoglobina fetale lega l'ossigeno più fortemente dell'emoglobina materna. In questo processo l'emoglobina fetale svolge un ruolo essenziale. L'ossigeno che attraversa la placenta e viene raccolto dall'emoglobina fetale è un elemento cruciale per la crescita del feto.
Il concetto chiave è che la maggiore affinità per l'ossigeno è dovuta alla ridotta interazione con il 2,3-BPG. Le subunità gamma dell'emoglobina fetale, rispetto alle subunità beta dell'emoglobina adulta, presentano meno amminoacidi basici positivi (mostrati in viola) nella regione di legame del 2,3-BPG. Questa differenza strutturale rende i siti di legame del 2,3-BPG meno basici e così impediscono la corretta interazione con il 2,3-BPG. Questo significa che il 2,3-BPG non si lega bene all'emoglobina fetale.
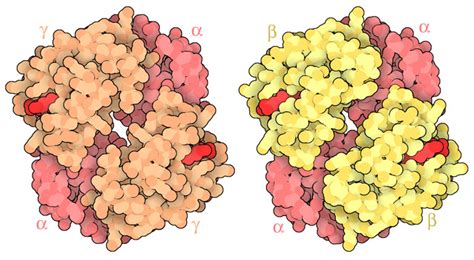
Emoglobinopatie: Variazioni e Implicazioni Cliniche
Un’emoglobinopatia è un disordine ematico ereditario caratterizzato dalla presenza di forme anomale dell’emoglobina (varianti emoglobiniche) o dalla riduzione della produzione della stessa (talassemia). L’emoglobina è formata da quattro subunità (globine o catene globiniche), ciascuna delle quali presenta al suo interno una struttura chiamata gruppo eme, contenente ferro e responsabile del legame dell’ossigeno. Esistono quattro tipi diversi di catene globiniche, designate come: α (alfa), β (beta), γ (gamma), δ (delta). La presenza di mutazioni nei geni codificanti per le catene globiniche può determinare la produzione di emoglobine anomale e quindi la presenza di emoglobinopatie. Queste mutazioni possono comportare la produzione di catene globiniche strutturalmente alterate o anche la perdita della produzione di uno o più tipi di catene globiniche.
Circa il 7% della popolazione mondiale è portatore in eterozigosi di una variante genetica in una delle catene dell’emoglobina; il tasso di mutazione può variare notevolmente in base all’etnia. Le mutazioni possono influenzare la struttura dell’emoglobina, la sua funzionalità, la sua velocità di produzione o anche la sua stabilità. La talassemia invece è causata dalla diminuita produzione o carenza di una delle catene globiniche. Esistono molteplici varianti emoglobiniche. Alcune silenti (ossia senza segni e sintomi evidenti) ed altre in grado di influenzare la funzionalità e/o la stabilità della molecola emoglobinica. Spesso, le forme meno comuni prendono il nome dal luogo di appartenenza della/e famiglia/e in cui la variante genetica è stata identificata per la prima volta.
Tipi Comuni di Emoglobinopatie
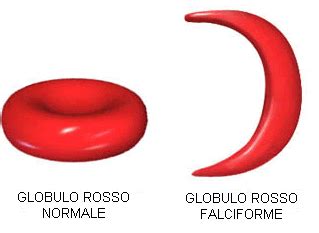
- Emoglobina S (HbS): Questa è responsabile dell’anemia falciforme. Si tratta di una patologia a frequenza molto variabile, ma comunque maggiormente presente nelle aree nelle quali la malaria è o è stata endemica, a causa di un vantaggio evolutivo dei portatori nei confronti della malaria. Le persone affette da anemia falciforme presentano entrambe le copie del gene responsabile della produzione di HbS, e pertanto presentano elevate quantità di HbS. Le persone con “tratto” falciforme (eterozigoti) presentano circa il 40% di HbS e il 60% di HbA normale. La presenza di HbS determina, in presenza di basse concentrazioni di ossigeno (come può accadere durante un esercizio fisico o in caso di infezioni polmonari), la variazione della forma dei globuli rossi che assumono la caratteristica forma a falce.
- Emoglobina C (HbC): Lo stato di portatore di HbC (eterozigoti) è presente in circa il 2-3% delle persone di origine africana. Lo stato di omozigosi comunque è un evento raro ed è responsabile di effetti relativamente lievi.
- Emoglobina E (HbE): È una delle varianti beta-globiniche più comuni nel mondo, con una maggiore prevalenza nelle persone originarie del sudest asiatico. I soggetti omozigoti per HbE in genere hanno una lieve anemia emolitica, macrocitosi ed un moderato ingrossamento della milza.
- Emoglobina H (HbH): Si forma in alcuni casi di alfa-talassemia. È composta da quattro catene beta globiniche (β4) e viene prodotta in caso di grave carenza delle catene alfa.
- Emoglobina di Bart (Hb Bart's): Si sviluppa nei feti affetti da alfa-talassemia. È composta da quattro catene globiniche gamma (γ4) e viene prodotta in caso di grave carenza delle catene alfa in maniera analoga a quanto avviene per l’HbH.
È possibile che la stessa persona erediti due differenti geni anomali, uno da ciascun genitore, con conseguente combinazione delle varianti emoglobiniche rilevate dai test. Questa condizione è nota come eterozigosi composta o doppia eterozigosi.
Diagnosi e Trattamento delle Emoglobinopatie
I test di primo livello per la ricerca delle emoglobinopatie in genere utilizzano metodiche volte alla determinazione del tipo e della quantità di emoglobine presenti nel sangue del paziente in esame (assetto emoglobinico). L’assetto emoglobinico si prefigge di rilevare eventuali varianti emoglobiniche e/o la quantità relativa dei diversi tipi di Hb. La maggior parte delle varianti emoglobiniche più comuni o delle talassemie possono essere identificate utilizzando una combinazione di questi test. La rilevazione della quantità relativa della variante emoglobinica presente è un valido ausilio diagnostico. Per la diagnosi delle emoglobinopatie l’esecuzione di un singolo test non è sufficiente. Questa richiede la valutazione complessiva di una serie di esami, eseguiti secondo processi ben definiti.
Mioglobina: funzioni e differenze rispetto all'Emoglobina 🩸
I test molecolari ricercano le mutazioni presenti nei geni codificanti per le catene globiniche alfa e beta. Il test per la ricerca delle emoglobinopatie viene richiesto come parte dei programmi di screening neonatale (attualmente attivo solo in alcune regioni in Italia). Lo screening delle emoglobinopatie consente di identificare e quindi potenzialmente anche trattare i neonati con disordini congeniti entro pochi giorni dalla nascita. In questo modo possono essere evitati problemi di salute potenzialmente letali o anche responsabili di disabilità permanenti. L’esame può essere richiesto nel caso in cui si sospetti un’emoglobinopatia in presenza di segni e sintomi caratteristici. Questo esame non viene eseguito in tutti i laboratori.
Nella valutazione delle emoglobinopatie deve essere prestata molta attenzione all’interpretazione dei risultati. I risultati in genere riportano il tipo di Hb presenti (quando identificabile) e la loro quantità relativa. Questo esame è un ausilio alla ricerca delle varianti emoglobiniche o di una classe di patologie note con il nome di talassemie, caratterizzate dalla diminuita produzione o carenza di una delle catene globiniche. Valori superiori alla norma dell’emoglobina A2 (oltre il 3,5% del totale) possono indicare la presenza di beta talassemia o anemia mediterranea.
Le trasfusioni di sangue possono interferire con la valutazione delle emoglobinopatie, poiché il metodo analitico non è in grado di distinguere tra l’emoglobina del donatore e quella del paziente, interferendo potenzialmente con i risultati del test. Un paziente dovrebbe aspettare diversi mesi dopo una trasfusione prima di sottoporsi a questo esame.
Il trattamento di alcuni tipi di emoglobinopatie può comportare l’utilizzo di terapie di supporto, ad esempio durante la comparsa di crisi in corso di anemia falciforme. L’obiettivo è quello di alleviare il dolore e di minimizzare le complicanze. Talvolta, in caso di anemia grave, è necessario ricorrere a trasfusioni di sangue. Meno frequentemente, possono essere utilizzati anche altri trattamenti.
Valori Normali dell'Emoglobina nel Sangue
Per gli adulti il valore dev'essere compreso tra 12 e 16 g/dL per le donne e tra 13,5 e 17 g/dL per gli uomini. Per i bambini può considerarsi normale il valore minimo di 10 g/dL. Questi valori sono indicativi per l'emoglobina adulta (HbA).
Strutture e Modelli Molecolari dell'Emoglobina e della Mioglobina
L'emoglobina e la mioglobina sono state soggetto di innumerevoli lavori di ricerca, che ne hanno svelato la complessa struttura tridimensionale. Questi studi sono stati fondamentali per comprendere i meccanismi molecolari alla base del legame e del rilascio dell'ossigeno. Le strutture di queste proteine sono state determinate attraverso tecniche avanzate, come la cristallografia a raggi X, e sono consultabili in database pubblici. Esempi di tali riferimenti includono i file PDB (Protein Data Bank) come 2h4z, 1b86 per l'emoglobina (Richard, V., Dodson, G. G., Mauguen, Y. complex low-salt structure at 2.5 A resolution. Journal of Molecular Biology), 1fdh per la deossiemoglobina (Frier, J. A., Perutz, M. F. deoxyhaemoglobin), e 4hhb per la struttura di deossiemoglobina umana ad alta risoluzione (Fermi, G., Perutz, M. F., Shaanan, B., Fourme, R. The crystal structure of human deoxyhaemoglobin at 1.74 Å resolution). Studi più recenti hanno approfondito i meccanismi molecolari coinvolti nella regolazione dell'emoglobina, come quelli che indagano BCL11A (Yang, Y., Xu, Z., He, C., Zhang, B., Shi, Y., Li, F. BCL11A) e l'efficacia di BCL11A per trattare l'anemia falciforme (Esrick, E. B., et al.). Altre ricerche hanno esaminato l'affinità dell'emoglobina fetale e adulta per il 2,3-Disfosfoglicerato (Tomita, S. fetal and adult hemoglobins by 2,3-Diphosphoglyceric Acid) o la nuova espressione placentare della 2,3-bisfosfoglicerato mutasi (Pritlove, D. C., Gu, M., Boyd, C. A. R., Randeva, H. S., Vatish, M. Novel placental expression of 2,3-bisphosphoglycerate mutase).
L'artista Julian Voss-Andreae ha creato nel 2005 una scultura dal titolo Cuore d'Acciaio (Heart of Steel), basata sulla struttura della proteina stessa. La scultura è realizzata in vetro e acciaio patinato. L'arrugginimento intenzionale dell'opera, inizialmente lucida, riflette la reazione chimica fondamentale dell'emoglobina, che lega l'ossigeno al ferro.

tags: #schema #che #riassume #emoglobina #mioglobina #emoglobina