L'emoglobina, una proteina fondamentale contenuta all'interno dei globuli rossi, svolge il ruolo cruciale di trasportare l'ossigeno ai tessuti del nostro organismo. La sua struttura è tipicamente un tetramero, formato da quattro catene proteiche. Un esempio eccellente di questa complessità genetica e funzionale è rappresentato dai geni che codificano per le catene che vanno a formare l'emoglobina, le cosiddette catene delle globine. Questi geni codificano per le catene α simili e β simili, che costituiscono le diverse forme di emoglobina presenti nelle diverse fasi dello sviluppo umano.
Questi geni delle globine costituiscono una famiglia genica, la cui trascrizione è finemente regolata in maniera differenziata nelle diverse fasi dello sviluppo. Questa regolazione è un processo non affatto semplice, ma essenziale per l'adattamento dell'organismo alle mutevoli esigenze di ossigenazione. Durante le prime otto settimane di gestazione, ad esempio, viene prodotta un'emoglobina embrionale specifica, caratterizzata dalla presenza di catene ζ (zeta) e ε (epsilon). Questa forma embrionale è ottimizzata per l'ambiente uterino precoce. Dopo l'ottava settimana, tuttavia, questa emoglobina scompare completamente. Durante questo periodo, avevano già iniziato a essere espressi i geni che codificano per le catene che troviamo nell'emoglobina fetale, prodotta da quel momento fino alla fine della gestazione. L'emoglobina fetale (HbF) α2γ2, con due catene alfa e due catene gamma, sostituisce dunque l'emoglobina embrionale e persiste per tutto il resto dello sviluppo intrauterino. In contrasto, le emoglobine di un adulto sono composte da due coppie di catene, che prendono il nome α (alfa) e ß (beta), costituendo l'emoglobina adulta (HbA), che è presente per il 98% con una struttura globulare.
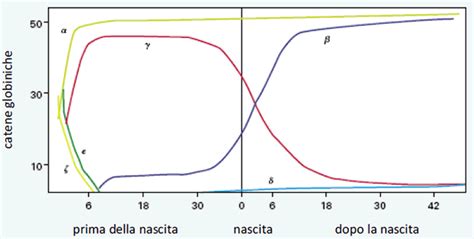
L'Emoglobina Fetale: Un Adattamento Cruciale per lo Sviluppo e il Trasporto di Ossigeno
Il passaggio dall'emoglobina embrionale a quella fetale, e successivamente a quella adulta, rappresenta un esempio lampante di come il controllo trascrizionale dei geni codificanti per proteine sia modulato in base alle esigenze fisiologiche dell'organismo in sviluppo. Nel contesto dello sviluppo fetale, l'emoglobina fetale (HbF) svolge un ruolo essenziale. L'emoglobina fetale, come menzionato, si distingue dall'emoglobina adulta (HbA) per la sua composizione, che prevede due catene α e due catene γ (gamma). Questa particolare struttura conferisce all'HbF proprietà uniche e fisiologicamente vantaggiose per il feto.
Una delle caratteristiche più importanti dell'emoglobina fetale è la sua maggiore affinità per l'ossigeno rispetto all'emoglobina adulta materna. Questa maggiore affinità è cruciale per il benessere del feto. A cosa serve l’emoglobina fetale in questo contesto? Riesce infatti a trasportare percentuali comprese tra il 20% e il 30% di ossigeno in più rispetto all'emoglobina materna. Questo meccanismo facilita il trasferimento di ossigeno attraverso la placenta. L'ossigeno, proveniente dal sangue materno e attraversando la placenta, viene efficacemente raccolto dall'emoglobina fetale presente nei globuli rossi del feto. Questa efficienza nel prelievo di ossigeno è dovuta, in parte, alla diversa interazione con il 2,3-bisfosfoglicerato (2,3-BPG).
Il 2,3-BPG è un metabolita che si lega all'emoglobina riducendone l'affinità per l'ossigeno, facilitandone così il rilascio nei tessuti. Tuttavia, l'emoglobina fetale lega il 2,3-BPG meno efficacemente rispetto all'emoglobina adulta. Specificatamente, i residui amminoacidici basici che si trovano nel centro della cavità dove si lega il 2,3-BPG nelle subunità beta dell'emoglobina adulta, sono sostituiti nelle subunità gamma dell'emoglobina fetale. Questa differenza nella composizione amminoacidica fa sì che l'emoglobina fetale leghi l'ossigeno più fortemente dell'emoglobina materna, che è influenzata in misura maggiore dal 2,3-BPG. Questa disparità di affinità assicura un gradiente di ossigeno favorevole, permettendo un efficiente passaggio di ossigeno dall'emoglobina adulta della madre a quella fetale. Questo è un perfetto esempio di adattamento biologico che garantisce la sopravvivenza e lo sviluppo ottimali del feto in un ambiente con livelli di ossigeno relativamente più bassi rispetto a quelli esterni.
EMOGLOBINA / LEZIONI DI BIOCHIMICA SUPER SEMPLIFICATE
Emoglobinopatie e il Ruolo Terapeutico dell'Emoglobina Fetale
Le emoglobinopatie sono un vasto gruppo di disordini genetici ereditari caratterizzati da varianti emoglobiniche o da una produzione insufficiente di emoglobina normale. Attualmente, sono state rilevate circa 1000 tipologie diverse di emoglobinopatie, che rappresentano un significativo problema di salute pubblica a livello globale. Tra queste, le più note e diffuse sono l'anemia falciforme e le talassemie, in particolare la beta-talassemia.
L'anemia falciforme è causata da una mutazione puntiforme nel gene della beta-globina, che porta alla produzione di una forma anomala di emoglobina, l'emoglobina S (HbS). In condizioni di bassa ossigenazione, l'HbS polimerizza e deforma i globuli rossi, facendoli piegare a forma di C o di falce. Questi globuli rossi "falciformi" sono rigidi, meno flessibili, e possono occludere i vasi sanguigni, causando crisi dolorose, danni agli organi e un'emolisi cronica. Le catene delle globine, che sono influenzate da questa mutazione, compromettono gravemente la funzionalità dei globuli rossi.
La beta-talassemia, invece, è caratterizzata da una ridotta o assente produzione di catene beta-globina, necessarie per la formazione dell'emoglobina adulta (HbA). Questo squilibrio porta a un eccesso di catene alfa-globina libere, le quali precipitano all'interno dei precursori eritroidi, causando un'eritropoiesi inefficace e un'anemia grave. Sia l'anemia falciforme sia la beta-talassemia sono malattie potenzialmente fatali, entrambe causate da mutazioni nella beta-globina, il gene che codifica per la molecola responsabile del trasporto dell'ossigeno.
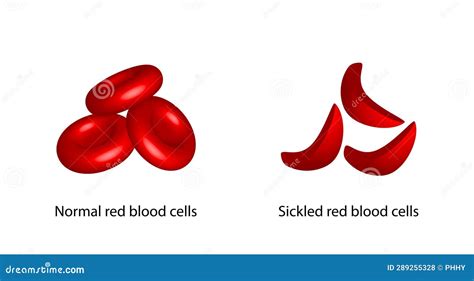
In queste patologie, l'emoglobina fetale emerge come un fattore protettivo di notevole importanza. È stato osservato che la presenza di livelli elevati di emoglobina fetale può mitigare significativamente la gravità dei sintomi. Ad esempio, è noto che circa il 30% di emoglobina fetale distribuito uniformemente nei globuli rossi allevia la maggior parte dei sintomi causati dall’anemia falciforme. Questo perché l'HbF non contiene le catene beta-globina mutate e non polimerizza come l'HbS, né è soggetta allo squilibrio delle catene che caratterizza la beta-talassemia. Nei pazienti affetti da anemia falciforme che mantengono una produzione di emoglobina fetale, manifestano la malattia in forma molto lieve, e in alcuni casi, livelli elevati di HbF nel sangue possono ridurre la frequenza e la gravità dei sintomi della malattia.
L'induzione della produzione dell’emoglobina fetale rappresenta dunque un potente approccio generalizzato per il trattamento dell’anemia falciforme e della maggior parte delle forme di beta talassemia, che possono essere causate da centinaia di mutazioni diverse. È proprio in questo contesto che la ricerca si è concentrata sulla comprensione e sulla manipolazione della regolazione genica dell'HbF per sviluppare nuove strategie terapeutiche.
La Persistenza Ereditaria dell'Emoglobina Fetale (HPFH): Un Modello Naturale di Protezione
Un fenomeno genetico di grande interesse clinico e scientifico è la Persistenza Ereditaria dell'Emoglobina Fetale (HPFH). Questa condizione è caratterizzata da livelli elevati di emoglobina fetale (HbF) nell'età adulta e da un aumento del numero delle cellule contenenti l'HbF, anche in assenza di stress emopoietico. La prevalenza di questa forma non è nota con precisione, data la sua eterogeneità genetica e la sua natura spesso asintomatica.
L'associazione tra la HPFH e condizioni come la beta-talassemia (BT) riveste un'importanza clinica straordinaria. Infatti, l'associazione tra la HPFH e la BT attenua i sintomi clinici nei pazienti, che possono essere asintomatici o presentare una beta-talassemia intermedia. In questi soggetti, pur avendo un difetto che elimina completamente la produzione di emoglobina beta adulta, la presenza continuativa dell'HbF compensa efficacemente la carenza di emoglobina adulta funzionale. Questa è una situazione di grande rilevanza, poiché negli altri soggetti con beta-talassemia, così come nei soggetti normali, l'emoglobina fetale viene completamente soppressa subito dopo la nascita, determinando il quadro clinico gravissimo della beta-talassemia maggiore, che richiede trasfusioni regolari di sangue per tutta la vita.
La HPFH è una condizione geneticamente eterogenea, dovuta a diverse alterazioni genetiche. Può derivare da delezioni nel cluster genico della beta-globina, che includono i geni HBG1 e HBG2 (localizzati sul cromosoma 11p15.5), o da mutazioni puntiformi all'interno dei promotori di questi stessi geni. Queste mutazioni impediscono la soppressione fisiologica dei geni della gamma-globina dopo la nascita.
La diagnosi di HPFH si basa sulla presenza di un significativo aumento dell'HbF nel sangue, che varia tipicamente tra il 10% e il 40% negli eterozigoti, con indici eritrocitari normali o quasi normali. Un aspetto distintivo è che l'HbF si distribuisce in maniera omogenea negli eritrociti. Inoltre, i livelli dell'emoglobina A2 (HbA2), una forma minore di emoglobina adulta, sono normali o ridotti nella HPFH. La differenziazione tra la HPFH e la delta-beta talassemia, un'altra condizione che può presentare livelli elevati di HbF, è spesso difficile e può essere confermata solo attraverso analisi specialistiche, come il rapporto di sintesi tra le globine alfa-beta e l'esame del DNA, poiché non sempre questa differenziazione è possibile sulla base dell'analisi standard del sangue. La trasmissione della HPFH è co-dominante, il che significa che un solo allele mutato è sufficiente per manifestare la condizione.
La comprensione della HPFH ha un impatto decisivo sulla cura della beta-talassemia, in quanto i meccanismi che portano alla persistenza dell'HbF rappresentano un modello naturale per lo sviluppo di terapie. L'obiettivo è riattivare la produzione di emoglobina fetale nella vita adulta, manipolando farmacologicamente o geneticamente i geni fetali per indurre una situazione simile alla HPFH, alleviando così i sintomi delle emoglobinopatie.
Regolazione Trascrizionale dei Geni Globinici e Approcci Terapeutici Innovativi
La regolazione genica dei geni globinici è un processo di straordinaria complessità, che si articola attraverso intricate reti di fattori di trascrizione e sequenze regolatorie, determinando quale tipo di emoglobina venga prodotta nelle diverse fasi della vita. Questa regolazione risponde alla domanda "Qual è il ruolo del controllo trascrizionale nei geni codificanti per proteine?" fornendo un esempio chiaro di come l'espressione genica sia finemente orchestrata. La domanda "Come si evolve l'emoglobina durante lo sviluppo embrionale e fetale?" trova risposta nell'osservazione della precisa sequenza di attivazione e silenziamento dei geni delle globine (ζ e ε nell'embrione, α e γ nel feto, α e β nell'adulto).
Nonostante l'identificazione di numerosi fattori di regolazione, finora i veri responsabili del controllo produttivo, soprattutto dei geni che producono emoglobina fetale, sono sfuggiti ai ricercatori. L'individuazione di questi geni avrebbe un impatto decisivo sulla cura della beta-talassemia, in quanto permetterebbe la manipolazione farmacologica o genetica dei geni fetali e la loro riattivazione nella vita adulta. Questa riattivazione condurrebbe a una situazione simile alla condizione clinica nota come persistenza ereditaria di emoglobina fetale (HPFH), offrendo una speranza significativa per i pazienti.
Progetti di ricerca attuali si propongono di individuare e caratterizzare i fattori di regolazione dei geni globinici. In questo contesto, sono state presentate nuove evidenze preliminari che indicano come alcuni fattori isolati dai ricercatori svolgano un ruolo di regolazione precedentemente sconosciuto sui geni globinici fetali e adulti. Questi fattori potrebbero, pertanto, essere utili nella terapia farmacologica o genetica della talassemia e dell'anemia falciforme, aprendo nuove vie per il trattamento di queste gravi condizioni.
La gestione clinica attuale della β-talassemia spesso impiega trasfusioni regolari di sangue per compensare l'anemia, terapia chelante per rimuovere l'eccesso di ferro accumulato a causa delle trasfusioni, e in alcuni casi, il trapianto di midollo osseo. Tuttavia, queste terapie sono gravose, non curative per tutti, e associate a significativi rischi e costi. Da qui l'esigenza di sviluppare trattamenti più efficaci e personalizzati, con un focus sempre maggiore sulla riattivazione dell'emoglobina fetale come strategia terapeutica. Le principali strategie per ripristinare la produzione di emoglobina (Hb) nei pazienti con β-talassemia si concentrano proprio sulla correzione della carenza o assenza di emoglobina adulta (HbA) o sull'induzione della produzione di emoglobina fetale (HbF), che può compensare la funzione dell'HbA.
EMOGLOBINA / LEZIONI DI BIOCHIMICA SUPER SEMPLIFICATE
Terapie Basate sull'Editing Genomico per le Emoglobinopatie
Le recenti scoperte nel campo dell'editing genomico hanno rivoluzionato le prospettive terapeutiche per le emoglobinopatie. L'editing genomico, in particolare il sistema CRISPR-Cas9, rappresenta un approccio molecolare molto efficace per correggere gli effetti delle mutazioni genetiche nelle malattie monogeniche ereditarie, inclusa la β-talassemia. Questo metodo consente di apportare modifiche precise al DNA, aprendo la strada a diverse strategie per il trattamento delle emoglobinopatie.
Correzione per la Produzione di Emoglobina Adulta (HbA) "de novo"
Un approccio diretto è la correzione basata sul gene editing (CRISPR-Cas9) per la produzione "de novo" di emoglobina adulta (HbA). Questo metodo consente di ottenere un'elevata produzione di HbA correggendo direttamente le mutazioni primarie del gene della β-globina. Sono stati riportati studi che dimostrano l'efficace correzione di varie mutazioni del gene della β-globina, tra cui HBB IVS2-654 (C > T), l'emoglobina E, il difetto di splicing della β654-talassemia, la mutazione stop codon β039-talassemia, la mutazione IVS-1-110 e la delezione β-41/42 (TCTT). L'obiettivo è ripristinare la funzionalità del gene della β-globina, consentendo la produzione di emoglobina adulta normale. Approcci più recenti come il base editing e il prime editing sono stati sviluppati per limitare la genotossicità associata alle rotture del doppio filamento del DNA (DSB) che avvengono con il CRISPR convenzionale, offrendo un'alternativa più sicura e precisa.
Induzione dell'Emoglobina Fetale (HbF) tramite Gene Editing
Un'altra strategia cruciale è l'induzione dell'emoglobina fetale (HbF) tramite gene editing (CRISPR-Cas9). L'aumento dell'HbF è clinicamente benefico per i pazienti con β-talassemia, potendo in alcuni casi portare all'indipendenza dalle trasfusioni, come visto nella HPFH. Questo obiettivo può essere raggiunto con CRISPR-Cas9 in due modi principali:
- Disruzione di geni che codificano per repressori trascrizionali: Questa tecnica mira a inattivare i geni che normalmente sopprimono l'espressione del gene della γ-globina (responsabile della produzione di HbF) dopo la nascita. Esempi includono BCL11A, SOX6 e KLF-1. La disruzione dell’enhancer di BCL11A, ad esempio, è stata associata alla riattivazione della produzione di HbF e all’indipendenza dalle trasfusioni in studi clinici, dimostrando l'efficacia di questo approccio. Il gene che codifica per l’emoglobina fetale è un ottimo bersaglio, dato che è sufficiente agire su un singolo nucleotide del DNA (una delle “lettere” del codice genetico) per riattivarlo, rendendolo un approccio molto studiato negli ultimi anni e ad oggi una strada promettente nella ricerca di una terapia per le emoglobinopatie.
- Disruzione dei siti di legame dei repressori nel promotore HBG: Questo approccio mima le mutazioni naturali della HPFH (persistenza ereditaria di emoglobina fetale), che sono associate a un decorso clinico più benigno nei pazienti con β-talassemia. Intervenendo sui siti specifici dove i fattori repressori si legano, si previene la repressione del gene della γ-globina, permettendone l'espressione continua. La disruzione del promotore del gene della β-globina adulta (HBB) può anche portare alla riattivazione dell’espressione del gene della γ-globina, in quanto il gene della γ-globina compete per il legame con il LCR (Locus Control Region), una regione regolatoria essenziale per l'espressione di tutti i geni globinici.
Riduzione dell'Eccesso di α-Globina Libera
Nella β-talassemia, l'eccesso di catene di α-globina libere è un fattore patofisiologico chiave che porta a eritropoiesi inefficace ed emolisi, contribuendo significativamente alla gravità della malattia. La riduzione di queste catene di α-globina ha un impatto clinicamente benefico. Il gene editing con CRISPR-Cas9 può essere utilizzato per ridurre l'espressione dei geni dell'α-globina, ad esempio mimando una mutazione naturale che causa α-talassemia o eliminando l'enhancer MCS-R2 dell'α-globina o il gene HBA2. Questo approccio complementare mira a ridurre la tossicità causata dall'accumulo di alfa-globina non accoppiata, migliorando l'efficacia dell'eritropoiesi.
Un team del St. Jude Children's Research Hospital e del Broad Institute del MIT e di Harvard ha messo a confronto cinque strategie di editing del genoma - utilizzando il più classico Cas9 o il più recente base editing - in cellule staminali ematopoietiche CD34+ e in cellule progenitrici. L’obiettivo è stato quello di valutare l’induzione della produzione dell’emoglobina fetale (HbF), in grado di sopperire alla mancanza dell’emoglobina che è tipica delle emoglobinopatie.
Il Base Editing e i Suoi Vantaggi nell'Induzione di HbF
Il base editing rappresenta una variante più recente e raffinata dell'editing genomico, distinguendosi dal classico CRISPR-Cas9 per la sua capacità di effettuare modifiche puntuali sulla sequenza di DNA senza indurre rotture del doppio filamento. Mentre CRISPR-Cas9 può tagliare, rimuovere o inserire segmenti di DNA nel filamento bersaglio, introducendo anche mutazioni puntiformi in una bassissima percentuale di casi, il base editing offre un controllo maggiore e una minore genotossicità. Questo lo rende un candidato promettente per trattamenti che richiedono precisione, anche se la sua sicurezza e efficacia necessitano di ulteriori valutazioni.
Nel contesto delle emoglobinopatie, il base editing ha dimostrato di essere particolarmente efficace per indurre la produzione di emoglobina fetale (HbF). Un gruppo di ricerca ha testato e confrontato gli effetti di Cas9 e di diversi base editor che agiscono sull’adenina. I risultati hanno rivelato che gli approcci di editing delle basi sono stati da 2 a 4 volte più potenti nell'aumentare l'espressione dell'emoglobina fetale, e hanno indotto livelli di HbF più stabili e più uniformi, rispetto all'uso del solo CRISPR-Cas9. Questa maggiore potenza e stabilità rendono il base editing una tecnologia attraente per la riattivazione dell'HbF, poiché un livello costante e ben distribuito di HbF è fondamentale per alleviare i sintomi delle malattie.
Inoltre, il base editing è stato utilizzato con successo per creare un nuovo sito di legame per un particolare fattore di trascrizione, denominato TAL1. Stando alle ricerche svolte dal gruppo, la creazione di questo sito indurrebbe una produzione maggiore di emoglobina fetale. Questa capacità di manipolare specifiche sequenze di legame per fattori trascrizionali apre nuove prospettive per modulare l'espressione genica dell'HbF in modo più mirato e potente.
Le emoglobinopatie, tra cui l'anemia falciforme e la beta talassemia, potrebbero beneficiare enormemente di questa modalità di intervento. L'obiettivo principale non è sempre quello di correggere l'errore genetico primario che scatena la malattia, ma piuttosto di compensare il difetto attraverso l'induzione dell'HbF. L'emoglobina fetale, infatti, non viene più prodotta dopo la nascita nella maggior parte degli individui, ma la sua riattivazione potrebbe essere la soluzione ideale per trattare le malattie causate da una carenza o assenza dell’emoglobina adulta funzionante. Questo è un approccio molto studiato negli ultimi anni e ad oggi pare essere una strada promettente nella ricerca di una terapia per le emoglobinopatie, offrendo una potenziale strada per migliorare la qualità di vita dei pazienti.
Induzione Farmacologica dell'Emoglobina Fetale: Un Approccio Complementare
Accanto alle innovative strategie di gene editing, l'induzione farmacologica dell'emoglobina fetale (HbF) attraverso molecole a basso peso molecolare costituisce un campo di ricerca in rapida evoluzione e un pilastro fondamentale nell'approccio terapeutico alle emoglobinopatie. L'idea è quella di utilizzare farmaci in grado di riattivare i geni della γ-globina negli adulti, mimando così la condizione protettiva della persistenza ereditaria dell'emoglobina fetale (HPFH).
Diversi induttori di HbF sono attualmente disponibili o in fase avanzata di studio clinico. Tra i più noti e utilizzati troviamo l'idrossiurea, un farmaco che agisce aumentando la produzione di HbF e che è già ampiamente impiegato nella gestione dell'anemia falciforme. Altri induttori in fase di studio includono il sirolimus (rapamicina), la talidomide e il 2,2-dimetilbutirrato (HQK-1001), ognuno con meccanismi d'azione specifici.
Questi agenti agiscono con meccanismi d'azione eterogenei, influenzando complessi percorsi di regolazione genica. Alcuni, ad esempio, inibiscono la metilazione del DNA, un processo epigenetico che porta al silenziamento genico. Altri agiscono inibendo le istone lisina metiltransferasi o l'attività delle istone deacetilasi (HDAC), enzimi coinvolti nella compattazione della cromatina e nella regolazione dell'espressione genica. Altri ancora attivano specifiche vie di segnalazione intracellulare, come la via p38 MAPK, o inibiscono la via mTOR, entrambe implicate nella differenziazione eritroide e nella modulazione della sintesi delle globine. Infine, alcuni induttori agiscono direttamente inibendo l'espressione dei repressori dei geni della γ-globina, contribuendo a mantenere attivo il programma fetale di produzione di emoglobina.
Un aspetto interessante di molti di questi induttori di HbF è che si tratta di farmaci riproposti o "ripocizionati" (repurposed drugs). Questo significa che sono farmaci già approvati per altre condizioni, la cui applicazione viene estesa alle emoglobinopatie una volta scoperti i loro effetti sull'HbF. Il riposizionamento di farmaci facilita notevolmente il loro trasferimento alla pratica clinica, accelerando i tempi di sviluppo e riducendo i costi associati alla scoperta di nuove molecole. Questa strategia farmacologica, da sola o in combinazione, si configura come un approccio promettente per la gestione a lungo termine delle emoglobinopatie.
Sinergie Terapeutiche: Combinare Approcci per Massimizzare i Benefici
L'efficacia e la sicurezza delle terapie per le emoglobinopatie possono essere massimizzate attraverso l'integrazione di diverse strategie, piuttosto che l'utilizzo di un singolo approccio. Per questo motivo, è stata proposta l'idea di combinare diverse strategie per massimizzare i benefici terapeutici e superare i limiti delle singole terapie, mirando a una cura più completa e duratura.
Combinazione di Gene Editing e Induzione Farmacologica di HbF
Una delle sinergie più promettenti è la combinazione di gene editing e induzione farmacologica di HbF. Questa strategia mira a ottenere la produzione di HbA "de novo" (tramite correzione del gene della β-globina) e, in parallelo, un aumento dell'HbF (tramite induttori farmacologici). L'obiettivo è duplice: ripristinare la produzione di emoglobina adulta funzionale dove possibile e, contemporaneamente, potenziare la produzione di emoglobina fetale come meccanismo compensatorio e protettivo. Uno studio ha dimostrato l'efficacia di questa combinazione utilizzando la correzione del gene β039-talassemia con CRISPR-Cas9 e l'induzione di HbF con rapamicina, ottenendo un aumento di entrambe le forme di emoglobina senza che una diminuisca l'altra. Questo approccio è particolarmente vantaggioso perché può minimizzare gli effetti collaterali (poiché il gene editing è singolo-plex, ovvero mira a una sola modifica) massimizzando al contempo la produzione totale di emoglobina, migliorando così significativamente il quadro clinico del paziente.
Combinazione di Gene Terapia e Gene Editing
Un'altra strada esplorata è la combinazione di gene terapia e gene editing. Alcuni approcci stanno esplorando l'utilizzo di vettori lentivirali (LV) che aggiungono un gene terapeutico, affiancato dall'editing genetico. Ad esempio, questa combinazione potrebbe essere utilizzata per ridurre la β-globina "sickling" (caratteristica dell'anemia falciforme) e indurre la globina anti-sickling, fornendo un doppio beneficio terapeutico. La gene terapia tradizionale introduce una copia funzionale del gene, mentre il gene editing corregge direttamente la mutazione o modula l'espressione genica endogena. L'integrazione di queste due metodologie potrebbe offrire soluzioni più robuste e complete.
Protocolli CRISPR-Cas9 Multiplex
Le ricerche menzionano anche la possibilità di utilizzare protocolli CRISPR-Cas9 multiplex, ovvero la capacità di effettuare più modifiche genetiche contemporaneamente. Ad esempio, si potrebbe silenziare il gene BCL11A e, al contempo, rompere i suoi siti di legame sul promotore del gene della γ-globina, combinando due strategie per l'induzione di HbF in un'unica procedura. Tuttavia, è necessario prestare cautela a causa della potenziale genotossicità e degli effetti off-target, inclusa la formazione di traslocazioni cromosomiche, che possono derivare da multiple rotture del DNA. Le nuove strategie di gene editing con minore genotossicità, come il base editing o il prime editing, potrebbero rendere questi approcci multiplex più sicuri e praticabili in futuro, riducendo i rischi associati all'instabilità genomica. In sintesi, la combinazione di diverse strategie terapeutiche rappresenta un orizzonte promettente per superare le complessità delle emoglobinopatie e offrire trattamenti personalizzati e altamente efficaci.
Implicazioni Cliniche e Prospettive Future: Il Caso di Exa-cel
Le prospettive terapeutiche per le emoglobinopatie stanno vivendo una fase di rapida evoluzione, con innovazioni che promettono di superare i limiti delle attuali gestioni cliniche, le quali spesso impiegano trasfusioni regolari di sangue, terapia chelante e, in casi selezionati, il trapianto di midollo osseo. Queste terapie, pur essendo salvavita, sono associate a una significativa morbilità e ridotta qualità di vita per i pazienti.
Un esempio emblematico di questa rivoluzione è rappresentato dall'avanzamento delle terapie basate sull'editing genomico. Un team del St. Jude Children's Research Hospital e del Broad Institute del MIT e di Harvard ha messo a confronto cinque strategie di editing del genoma - utilizzando il più classico Cas9 o il più recente base editing - in cellule staminali ematopoietiche CD34+ e in cellule progenitrici. L’obiettivo primario di questa ricerca è stato quello di valutare l’induzione della produzione dell’emoglobina fetale (HbF), in grado di sopperire alla mancanza dell’emoglobina adulta che è tipica delle emoglobinopatie.
Nel contesto dei trattamenti in fase di studio per queste patologie, la terapia genica ha già mostrato notevoli successi. Per la beta-talassemia, una terapia genica ha ottenuto l'approvazione, ma non è più disponibile sul mercato europeo da maggio 2022, sottolineando la dinamicità e le sfide regolatorie in questo settore.
Nel frattempo, la prima terapia basata su CRISPR arrivata alla sperimentazione sull’uomo è proprio per una emoglobinopatia. Exagamglogene autotemcel (exa-cel) è stata una delle prime terapie a base di CRISPR ad entrare in trial clinico circa quattro anni fa, per trattare un caso di anemia falciforme. La storia di successo di Victoria Gray, la prima paziente trattata e "curata" con CRISPR, è diventata un simbolo delle potenzialità di questa tecnologia. Un segnale della sua imminente disponibilità su larga scala è l'accoglimento, avvenuto a febbraio, della domanda di autorizzazione all’immissione in commercio di exa-cel da parte dell’Agenzia Europea dei Medicinali.
CTX001, il nome clinico per le terapie exa-cel e beti-cel (per la beta-talassemia), rappresenta una terapia che corregge il difetto a livello genico con la tecnica CRISPR/Cas9. Questa metodologia è mirata a ingegnerizzare le cellule staminali del midollo osseo per indurle a produrre alte quantità di emoglobina fetale (HbF), un'emoglobina presente normalmente nel feto e nei primi mesi di vita e che può efficacemente compensare il difetto di queste malattie. La correzione viene effettuata in vitro su cellule staminali ematopoietiche, prelevate dal midollo osseo del malato. Queste cellule, una volta modificate geneticamente e destinate a diventare progenitrici dei globuli rossi, vengono poi reiniettate nel paziente attraverso un catetere venoso centrale. L'intento è quello di ripopolare il midollo di quel paziente con progenitori di globuli rossi capaci di produrre HbF. Queste staminali geneticamente modificate e localizzate nel midollo maturano fino a diventare globuli rossi corretti contenenti emoglobina fetale, adeguata efficacemente al trasporto di ossigeno.
EMOGLOBINA / LEZIONI DI BIOCHIMICA SUPER SEMPLIFICATE
I trial clinici programmati prevedono il trattamento di circa una quarantina di pazienti, con l'obiettivo primario di indagare la sicurezza e l'efficacia della terapia. In particolare, l'efficacia sarà valutata in base alla diminuzione del numero di trasfusioni di sangue, che sono indispensabili con notevole frequenza nelle forme gravi di tali malattie. Questa osservazione porta a importanti considerazioni su possibili approcci analoghi per il trattamento di altri difetti genetici, sebbene l'approccio con gene editing sia tecnicamente più semplice nelle anemie genetiche rispetto ad altre condizioni, come ad esempio la fibrosi cistica, dove la complessità degli organi coinvolti e la via di somministrazione delle staminali ingegnerizzate pongono sfide maggiori. L'avanzamento delle terapie basate sull'editing genomico promette dunque di trasformare radicalmente il panorama terapeutico delle emoglobinopatie, offrendo una speranza concreta per una cura funzionale.
Monitoraggio e Diagnosi: L'Esame dell'Emoglobina Fetale
Oltre al suo ruolo cruciale nello sviluppo e nelle nuove strategie terapeutiche, l'emoglobina fetale (HbF) è anche un importante biomarcatore diagnostico. L'esame per l'emoglobina fetale rientra tra i diversi esami del sangue svolti per la diagnosi e il monitoraggio di patologie a carico del sangue, in particolare talassemia e anemia falciforme. La sua misurazione è essenziale per comprendere lo stato della malattia e per orientare le decisioni cliniche.
I livelli di emoglobina fetale nell'adulto sono normalmente molto bassi. In un adulto, livelli di emoglobina fetale considerabili normali oscillano tipicamente tra 0.1 e 1.1%. Tuttavia, è fondamentale ricordare che i valori di riferimento degli esami di laboratorio possono variare a seconda della metodologia di analisi dei campioni e del laboratorio specifico; quelli indicati hanno uno scopo puramente informativo e devono essere interpretati da un medico.
La domanda "Emoglobina fetale, quando preoccuparsi?" si pone quando negli adulti si riscontrano valori superiori a 1.1%. In questi casi, si parla di emoglobina f alta. Un innalzamento significativo dei livelli di HbF può essere indicativo di diverse condizioni. Oltre alle emoglobinopatie come la beta-talassemia e l'anemia falciforme, dove l'HbF alta può essere un meccanismo compensatorio naturale, può anche indicare una condizione di recupero dovuta a ipoplasia di midollo osseo, ovvero un disturbo delle cellule staminali ematopoietiche. In questi scenari, il corpo tenta di compensare la produzione di globuli rossi insufficiente riattivando la produzione di emoglobina fetale, che è un indicatore di stress eritropoietico o di una risposta adattativa.
Per l'esecuzione dell'esame dell'emoglobina fetale, come per molti altri esami ematici, è generalmente richiesto un digiuno di almeno otto ore. Questa preparazione standard assicura che i risultati non siano influenzati da recenti assunzioni di cibo o bevande, garantendo la massima accuratezza e affidabilità del test. La corretta interpretazione dei livelli di HbF, in combinazione con altri parametri ematologici e la storia clinica del paziente, è quindi cruciale per una diagnosi precisa e per la gestione ottimale delle patologie del sangue.

Oltre il DNA: L'Editing dell'RNA Messaggero e Nuove Frontiere Terapeutiche
Il campo della terapia genica e dell'editing genomico è in costante evoluzione, e la ricerca non si limita alla manipolazione diretta del DNA. In linea generale, l'approccio con gene editing è tecnicamente più semplice nelle anemie genetiche rispetto a patologie più complesse come la fibrosi cistica (FC), dove, tra le altre cose, è ancora da scoprire per quale via sarebbe possibile un'analoga somministrazione di staminali ingegnerizzate per correggere le mutazioni CFTR implicate. Tuttavia, l'innovazione non si ferma e si esplorano nuove strade per superare queste sfide.
Una strada promettente è la correzione, piuttosto che del DNA del gene, del suo RNA messaggero (RNAm). L'RNAm è l'acido nucleico che trasmette le informazioni contenute nel DNA genico, al fine di produrre la specifica proteina di cui è responsabile. Se un gene è mutato, il suo DNA ha una sequenza alterata, il che dà origine a un RNAm alterato e, di conseguenza, a una proteina difettosa. L'idea è che anche questo RNAm possa essere corretto, potenzialmente sempre attraverso una variante della tecnica CRISPR, come dimostrato in alcuni contesti di ricerca. In questo modo ci sarebbe il vantaggio di non manipolare direttamente il DNA, evitando così potenziali effetti di lunga durata o effetti off-target su altri geni nel genoma, il che potrebbe offrire un profilo di sicurezza più favorevole.
Per far capire quanto l'argomento sia in fermento e quanto siano veloci gli avanzamenti, citiamo che, a livello di grandi industrie farmaceutiche, mentre CRISPR Therapeutics, menzionata in precedenza per le sue ricerche sulla correzione del DNA, si concentra su approcci diretti al genoma, un'altra azienda, Translate Bio (Lexington, Massachusetts), punta proprio sull'RNAm. Questa azienda sta lavorando in collaborazione con ricercatori del prestigioso MIT (Massachusetts Institute of Technology) per sviluppare terapie basate sull'RNA messaggero.
In questo caso, l'approccio non sfrutta direttamente il sistema CRISPR sul DNA. L'idea è di aggirare il problema del DNA alterato e tentare di introdurre nella cellula RNA messaggero con una sequenza normale. Questo RNAm esogeno, una volta all'interno della cellula, è dotato delle istruzioni corrette per produrre la proteina in questione, bypassando il gene difettoso nel nucleo. Attraverso esperimenti su polmone di topi, trattati per via aerosolica, i ricercatori hanno dimostrato con successo la possibilità di introdurre nelle cellule epiteliali un RNA messaggero destinato alla sintesi dell’enzima luciferasi (l’enzima che permette alle lucciole di fare luce). Questo esperimento di prova di concetto apre la strada all'applicazione di tale tecnologia per introdurre RNAm terapeutici in diverse patologie, comprese le emoglobinopatie o altre malattie genetiche, offrendo un'alternativa intrigante all'editing del DNA con implicazioni potenzialmente meno invasive e più reversibili.
tags: #regolazione #genica #emoglobina #fetale