La crescita è un processo complesso e delicato, fondamentale per lo sviluppo armonico di ogni bambino. Quando si verificano disordini della crescita, l'intero nucleo familiare è coinvolto in un percorso spesso lungo e impegnativo. Il progetto web GrowingUp è nato proprio per offrire supporto alle famiglie di bambini affetti da disordini della crescita, realizzato con il contributo di esperti e in collaborazione con genitori di bambini che stanno affrontando la patologia. Si propone di portare un’informazione chiara, completa, aggiornata e consigli pratici per tutta la famiglia coinvolta. Un disordine della crescita è qualsiasi tipo di problema nei bambini, ragazzi o adolescenti che impedisca di raggiungere il livello di crescita atteso secondo i normali parametri. Tra le cause più significative si annoverano le problematiche legate all'ipofisi, una piccola ghiandola situata alla base del cervello, cruciale per la produzione di numerosi ormoni essenziali.
Comprendere i Disordini della Crescita e l'Ipopituitarismo
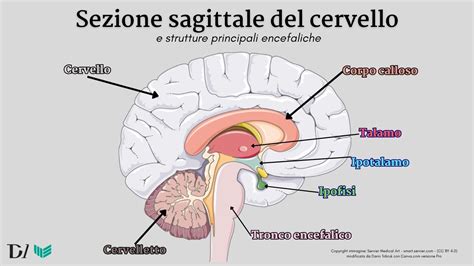
L'ipofisi, o ghiandola pituitaria, è una ghiandola che si trova alla base del cervello e produce l’ormone della crescita, regolando la crescita e lo sviluppo fisico. Talvolta, però, sono altri gli ormoni pituitari a mancare, e in tal caso si parla di ipopituitarismo o insufficienza ipofisaria. Se l’ipofisi non produce ormone della crescita in quantità sufficiente, la crescita può risultare rallentata, con conseguente bassa statura. Questo è conosciuto come Deficit di ormone della crescita (GH), una patologia che ad oggi colpisce in Italia 1 bambino ogni 3.000 nati vivi. È il più comune deficit di ormoni ipofisari ed è accompagnato da una scarsa crescita complessiva e da bassa statura. I bambini con deficit di ormone della crescita possono essere anche carenti in altri ormoni ipofisari, come l'ormone tireostimolante, l'ormone adrenocorticotropo, l'ormone follicolo-stimolante e l'ormone luteinizzante. Un disturbo che coinvolge la carenza di multipli ormoni ipofisari viene chiamato ipopituitarismo.
L'ipopituitarismo congenito è caratterizzato da deficit multiplo degli ormoni ipofisari, come il deficit dell'ormone somatotropo, tireotropo, lattotropo, corticotropo o gonadotropo, causato da mutazioni dei fattori di trascrizione coinvolti nell'ontogenesi dell'ipofisi. L'incidenza stimata dell'ipopituitarismo congenito è compresa tra 1/3000 e 1/4000 nascite, rientrando ampiamente nella categoria delle malattie rare. Un bambino affetto da Deficit di ormone della crescita manifesta una crescita di meno di 5 centimetri all’anno. In molti casi la crescita è regolare sino al secondo o terzo anno di età e mostra segni di rallentamento dopo i tre anni. In altri casi, però, la mancata crescita si manifesta prima o dopo queste soglie di età. L'assenza di crescita può portare a quello che viene chiamato comunemente "nanismo ipofisario" se la mancanza è precoce. Si tratta di bambini di bassa statura e un aspetto particolare del volto - viene indicato come aspetto a facies di bambola -, cioè bambini che sembrano più piccoli di quanto siano in realtà e che, se non trattati in maniera corretta, possono avere delle caratteristiche che possono arrivare all'aspetto vecchieggiante della cute. Il campanello d'allarme più importante è la velocità di crescita. Analogamente a come misuriamo i chilometri orari di un'auto, possiamo misurare i centimetri annui di crescita di un bambino. Se in base a delle tabelle definite, un bambino presenta una velocità di crescita normale, significa non solo che non può essere affetto da questo deficit ma che sta bene perché la velocità di crescita è l'indice più importante del benessere di un bambino.
Panipopituitarismo nel Lattante: Sintomi e Segni Clinici
La presentazione clinica del panipopituitarismo nel lattante può essere estremamente variabile, rendendo la diagnosi una vera e propria sfida per il clinico. La sintomatologia con cui può presentarsi il deficit ormonale multiplo è variabile in base al tipo ed alla severità del deficit combinato. I sintomi di deficit di ormone della crescita dipendono dall'età del bambino e dalla causa della carenza. Nello specifico, i neonati con deficit di ormone della crescita possono avere bassi livelli di zucchero nel sangue (ipoglicemia), ittero (iperbilirubinemia) o altre anomalie congenite come un pene piccolo (micropenia) nei maschi o difetti facciali (come la palatoschisi).
Screening Neonatale - Intervista alla dott.ssa Spina
In epoca neonatale, segni e sintomi da attenzionare includono ittero prolungato, ipoglicemia persistente e severa in assenza di iperinsulinismo, ipotonia, difficoltà ad alimentarsi, ipo/ipernatriemia, micropene e/o criptorchidismo. L'ipoglicemia severa e protratta è il più importante e comune sintomo di presentazione del panipopituitarismo neonatale ed è causata dal deficit degli ormoni controinsulari GH ed ACTH. L’ipoglicemia può essere associata ad iponatremia e ad instabilità cardiocircolatoria che può esitare in shock in caso di evento stressante a causa del deficit dell’asse corticotropo. Il sintomo ipoglicemia può essere misconosciuto in epoca neonatale, specie nel prematuro in prolungata nutrizione parenterale.
Nei maschi affetti, una caratteristica del panipopituitarismo neonatale è la presenza di micropene (lunghezza del pene inferiore alle -2,5 SDS) nel 60% dei casi. Possono essere evidenti anche i caratteri fenotipici legati al deficit di GH, come bozze frontali prominenti, naso a sella, ipotonia della muscolatura addominale con possibile ernia ombelicale. Nei casi associati ad anomalie della linea mediana, è possibile riscontrare labio-palatoschisi, incisivo singolo centrale ed ipertelorismo. L’ovvia mancanza di quest’ultimo segno nelle bambine può ritardare il sospetto diagnostico. La bassa statura corrisponde a un’altezza inferiore al terzo percentile dell’età del bambino (in base alle curve di crescita standard per età e altezza). I bambini presentano tassi di crescita complessivi scarsi. Di solito crescono meno di 6 centimetri all’anno prima dei 4 anni, meno di 5 centimetri all’anno dai 4 agli 8 anni e meno di 4 centimetri all’anno prima della pubertà. La maggior parte presenta bassa statura, ma proporzioni normali del corpo superiore e inferiore. In alcuni casi si può verificare un ritardo nello sviluppo della dentizione.
Le Cause del Panipopituitarismo Congenito
Le cause del deficit di ormone della crescita e dell'ipopituitarismo possono essere suddivise in congenite e acquisite. Nella maggioranza dei casi di deficit di ormone della crescita, la causa è sconosciuta, ma a volte può trattarsi di una malattia congenita o di un tumore cerebrale. Le forme acquisite di deficit di GH, in genere, sono legate a problemi sulla struttura dell'ipofisi. Ci possono essere dei tumori, anche se benigni, che possono determinare un malfunzionamento della ghiandola ipofisaria. Altre cause acquisite includono infezioni (come la meningite) o traumi, e la radioterapia può aumentare il rischio di ipopituitarismo.
Per quanto riguarda le forme congenite, l'ipopituitarismo congenito è dovuto a una insufficiente produzione di uno o più ormoni prodotti dall’adenoipofisi, associata o meno a un’insufficiente secrezione di ADH da parte della neuroipofisi. Nella maggior parte dei casi pediatrici l’ipopituitarismo è dovuto a un’anomalia nel processo di formazione e sviluppo dell’ipofisi. Questa anomalia può essere causata da noxae patogene (infezioni, abuso di sostanze voluttuarie, inalazione di agenti tossici atmosferici) occorse in varie fasi della gravidanza.

Più frequentemente, l'ipopituitarismo congenito è legato a mutazioni a carico dei geni che regolano il normale processo di formazione e sviluppo della linea mediana. L’ipopituitarismo congenito è causato da mutazioni di diversi geni che codificano per i fattori di trascrizione. Il fenotipo varia a seconda del fattore di trascrizione coinvolto:
- PROP1: Deficit degli ormoni somatolattotropo, tireotropo, gonadotropo e a volte corticotropo; iperplasia e ipoplasia ipofisaria. Questi pazienti possono sviluppare nuove carenze (per esempio un esordio tardivo di deficit dell'ormone corticotropo). La trasmissione è recessiva.
- POU1F1: Carenza degli ormoni somatolattotropo e tireotropo, ipoplasia ipofisaria. La trasmissione è autosomica recessiva.
- HESX1: Carenze ipofisarie variabili, displasia setto-ottica. La trasmissione è autosomica recessiva.
- LHX3: Deficit degli ormoni somatolattotropo e gonadotropo, limitazioni nella rotazione del capo e del collo. La trasmissione è recessiva.
- LHX4: Deficit ipofisari variabili, ectopia della neuroipofisi, anomalie dell'encefalo. La trasmissione è dominante.
L’ipopituitarismo congenito, inoltre, può essere parte di forme sindromiche come nella displasia setto-ottica. Esistono delle malattie genetiche per cui alcuni geni non permettono la formazione normale dell'ipofisi e si ha un deficit strutturale dell'ipofisi definito ipopituitarismo congenito che può essere evidente già in fase neonatale.
Processo Diagnostico: Dalla Valutazione Clinica ai Test Specifici
La diagnosi di ipopituitarismo, specialmente in epoca neonatale, è complessa a causa della sintomatologia variabile e spesso aspecifica. La diagnosi si basa sulla combinazione di segni clinici, dati di laboratorio, imaging e test genetici. Il pediatra o l’endocrinologo cercano i segni delle diverse possibili cause della mancata crescita o di una bassa statura.Per prima cosa, i medici misurano la statura e il peso del bambino rapportandoli alle curve di crescita in base all’età, per stabilire se il bambino ha una crescita ridotta. Per la diagnosi si basa su esame obiettivo, revisione delle curve di crescita del bambino e da analisi che possono comprendere radiografie, esami del sangue, esami genetici, test di stimolazione ed esami di diagnostica per immagini.
Valutazione Clinica e Strumentale:
- Anamnesi e Esame Obiettivo: Valutazione medica dei parametri di crescita e anamnesi dei disturbi riconosciuti come causa del rallentamento della crescita.
- Radiografie: Spesso si esegue la radiografia delle ossa della mano (radiografia per la valutazione dell’età ossea), che rivela se il loro sviluppo è nella norma rispetto all’età del bambino. I bambini semplicemente bassi di statura hanno crescita ossea normale per la loro età. Quelli con deficit dell’ormone della crescita presentano ritardi dello sviluppo osseo, anche se ciò può accadere anche per altre condizioni, come ipotiroidismo e ritardo della pubertà.
Esami di Laboratorio:
- Test Ematici Ormonali: Ai fini della corretta diagnosi possono richiedere test ematici per valutare ormoni e anomalie nei cromosomi. È importante escludere tutte le patologie che possono dare rallentamento della capacità di crescita o bassa statura per evitare un eccesso di diagnosi. Con gli esami del sangue i medici misurano i livelli di altre sostanze presenti nel sangue che sono stimolate dall’ormone della crescita, come il fattore di crescita insulino-simile 1 (IGF-1) e la proteina legante il fattore di crescita insulino-simile 3 (IGFBP-3). Tuttavia, queste sostanze possono essere influenzate da altre condizioni, come ipotiroidismo, celiachia e denutrizione, pertanto i medici conducono test per escludere queste condizioni. Si eseguono anche esami di laboratorio alla ricerca di altre cause della scarsa crescita (ad esempio disturbi di tiroide, sangue, reni, infiammatori e del sistema immunitario).
- Test di Stimolazione dell'Ormone della Crescita: Per valutare la capacità dell’ipofisi del bambino di produrre ormone della crescita, il medico - di solito l’endocrinologo pediatrico - può richiedere il test di stimolazione dell’ormone della crescita. Se il bambino non presenta altre cause per la scarsa crescita e i livelli dell’ormone della crescita sono bassi, i medici di solito eseguono un test di stimolazione, che prevede la somministrazione di farmaci che stimolano la produzione dell’ormone della crescita, misurandone quindi i livelli nel corso di varie ore. I livelli dell’ormone della crescita nel sangue sono molto variabili e non sono utili quanto altri livelli ormonali per determinare la causa del calo della crescita di un bambino. Pertanto, i medici fanno la diagnosi basandosi sui riscontri disponibili.
- Valutazione dell'Asse Tiroideo e Gonadico: È cruciale la valutazione dell’asse tiroideo e gonadico, con riscontro di FT4 e TSH, e di FSH e LH, come parte della diagnosi di ipopituitarismo congenito. La valutazione ormonale, in particolare il dosaggio di ACTH, cortisolo e GH in corso di ipoglicemia, è dirimente.
Imaging e Genetica:
- Risonanza Magnetica (RM) Encefalo: Se il test suggerisce che il bambino è affetto da un disturbo dell’ipofisi, si può procedere con esami di diagnostica per immagini del cervello mediante risonanza magnetica per immagini (RMI) per ricercare anomalie strutturali dell’ipofisi e tumori. La valutazione neuroradiologica e l’analisi dei geni coinvolti nell’ontogenesi della regione ipotalamo-ipofisaria sono fondamentali per la diagnosi e per comprendere lo sviluppo embriologico di questa regione cardine in ambito ormonale.
- Test Genetici: I test genetici possono essere fatti se i medici temono che il bambino sia affetto da una sindrome specifica (come la sindrome di Turner). La conferma della diagnosi avviene tramite sequenziamento diretto dei geni dei fattori di trascrizione. Un screening clinico, biologico e radiologico è fondamentale per determinare al meglio quale fattore di trascrizione dovrebbe essere indagato.
La Terapia Farmacologica: Ormoni Sostitutivi e Gestione
La terapia del panipopituitarismo è di tipo ormonale sostitutivo, con modulazione in base alla risposta clinica e ai parametri di laboratorio. Se si identifica una condizione medica specifica, il trattamento adeguato può portare ad una crescita sostanzialmente nella norma. È necessaria un'appropriata integrazione delle carenze ormonali. Ai bambini affetti da ipopituitarismo vengono somministrati ormoni per compensare quelli mancanti.
Terapia con Ormone della Crescita (GH):Si somministra l'ormone della crescita. Il trattamento generalmente consiste nell’ormone tiroideo sostitutivo. Al bambino vengono somministrate iniezioni di ormone della crescita sintetico. Le iniezioni vengono tradizionalmente somministrate una volta al giorno, ma formulazioni più recenti di ormone della crescita vengono iniettate una volta alla settimana, come si sta sperimentando al Bambino Gesù con un prodotto che ha un'efficacia che si prolunga per sette giorni così da non dover fare iniezioni quotidiane. La terapia continua fino al raggiungimento della statura definitiva e qualche volta deve continuare anche nella vita adulta quando il difetto è molto significativo tanto da dare problemi a livello muscolare/metabolico. Durante il primo anno di trattamento il bambino può crescere fino a 10-12 cm, ma la risposta è soggettiva. Si tratta di un farmaco riconosciuto dal Servizio Sanitario Nazionale se rientra nelle caratteristiche della Nota 39, un sistema di controllo dell'appropriatezza della terapia. Si può dare solo nelle condizioni specifiche indicate dalla Nota 39. Quando non ci sono, non può essere dato e non deve essere dato.
Altri Ormoni Sostitutivi:
- L-Tiroxina: Per la carenza di ormone tireostimolante (TSH) o ipotiroidismo centrale.
- Idrocortisone: Per la carenza di ormone adrenocorticotropo (ACTH) o insufficienza surrenalica. Dal punto di vista clinico, la gestione della terapia sostitutiva implica che è fondamentale correggere per primo il deficit di cortisolo, un ormone indispensabile per la vita soprattutto in condizioni di stress.
- Analoghi del GnRH: La pubertà precoce, caratterizzata dall’insorgenza anticipata dei segni di maturazione sessuale, può influire negativamente sulla crescita e sul benessere psicologico del bambino. Gli analoghi del GnRH sono farmaci che agiscono bloccando temporaneamente l’attivazione dell’asse ipotalamo-ipofisi-gonadi. La terapia con analoghi del GnRH è indicata principalmente nei casi di pubertà precoce centrale, ovvero quando l’asse ormonale si attiva troppo presto e in modo completo. La terapia con analoghi del GnRH è generalmente ben tollerata ed è uno strumento fondamentale per gestire la pubertà precoce centrale, preservando la crescita e il benessere del bambino.
Considerazioni sul Trattamento:
- L’analisi delle evidenze emerse dalle 181 testimonianze ha portato alla luce alcune criticità che possono mettere a rischio l’aderenza alla terapia, e quindi l’efficacia della stessa.
- Solitamente la terapia a base di ormone della crescita non provoca effetti collaterali nei bambini, sebbene alcuni sviluppino un lieve gonfiore degli arti che in genere si risolve rapidamente oppure, raramente, effetti collaterali più gravi, come aumento della pressione cerebrale (ipertensione intracranica idiopatica) o un problema nella parte superiore del femore, che può manifestarsi come dolore al ginocchio o all’anca oppure andatura zoppicante (slittamento dell’epifisi femorale capitale).
Casi Clinici e Sfide Diagnostiche
Il sospetto e la diagnosi di ipopituitarismo congenito rappresentano una sfida per il clinico, soprattutto in ambito neonatale, a causa della sintomatologia variabile e spesso aspecifica. Il panipopituitarismo neonatale merita un’attenzione particolare dato che il suo mancato riconoscimento può esitare in complicanze acute che mettono a rischio la vita del neonato; nei casi meno gravi può invece determinare un deficit accrescitivo ed un inadeguato sviluppo neuromotorio.
Un esempio di questa complessità è il caso di un lattante nato a 40+4 settimane di gestazione da TC urgente, con una gravidanza caratterizzata da diabete e ipotiroidismo gestazionale. Alla nascita, si è riscontrata necessità di ventilazione e glicemia bassa, che ha richiesto infusione con soluzione glucosata al 10%. Nel sospetto di ipopituitarismo congenito, la valutazione dell’asse tiroideo e gonadico ha mostrato un FT4 basso con TSH inappropriatamente basso e FSH/LH ridotti rispetto alla fisiologica attivazione neonatale. Clinicamente, il piccolo presentava scarso accrescimento in peso e lunghezza, ipotono, iporeattività con difficoltà nella suzione e subittero. La RM encefalo ha rivelato un quadro di displasia setto-ottica con cavo sellare di piccole dimensioni, aspetto ipoplasico dell’adenoipofisi, peduncolo ipofisario presente ma assottigliato ed ectopia della neuroipofisi, oltre ad asimmetria dei nervi ottici e corpo calloso sottile. È stata avviata terapia ormonale sostitutiva con idrocortisone, L-tiroxina e GH, dopo la quale si è osservato un miglioramento delle condizioni cliniche generali e non si sono più verificati episodi di ipoglicemia. L'indagine genetica è ancora in corso.
Un altro caso significativo è quello di Martina, nata con parto distocico e successivamente ricoverata per crisi ipoglicemiche ricorrenti. A 6 mesi, ha avuto due gravi crisi ipoglicemiche, portando a una diagnosi iniziale di malattia del metabolismo degli aminoacidi e alimentazione con sondino naso-gastrico. Martina ha mostrato un ritardo nello sviluppo psico-motorio ed è stata seguita per sospetta malattia metabolica fino all'età di 7 anni, con pasti frazionati per prevenire le crisi ipoglicemiche. Solo in seguito è stato riscontrato un ipotiroidismo centrale, per cui è stata iniziata terapia con L-tiroxina, che ha causato scompenso della paziente. Successivamente, è stato diagnosticato un grave deficit surrenalico e di ormone della crescita, che, unitamente all’ipotiroidismo centrale, sono diagnostici per un panipopituitarismo. Una RM della regione ipotalamo-ipofisi ha dimostrato la presenza di una sella vuota con neuroipofisi ectopica, confermando il panipopituitarismo. La bambina è stata messa in trattamento con idrocortisone unitamente a L-tiroxina, e successivamente terapia con GH ricombinante. Il trattamento sostitutivo ha determinato un miglioramento delle condizioni della bambina, un incremento della velocità di crescita e un progressivo miglioramento delle funzioni cognitive. La storia clinica di Martina dimostra come, a volte, un panipopituitarismo possa rimanere una diagnosi difficile anche in presenza di una storia clinica e una sintomatologia tipica, risultandone una tardiva individuazione con possibili ripercussioni sia fisiche che neuro-cognitive. L’assenza di un grave deficit accrescitivo, da mettere in relazione con l’iperalimentazione a cui è stata sottoposta la bambina fino a 7 anni per prevenire le crisi ipoglicemiche inducendo uno stato di obesità, può avere contribuito al ritardo diagnostico.
Considerazioni sull'Uso dell'Ormone della Crescita: Efficacia, Rischi e Controversie
L’ormone della crescita (GH) ha un bel nome, pieno, accattivante, convincente, che fa pensare che esista una cosa ben precisa che fa diventare grandi, o, almeno, più alti. Per questo la proposta di un farmaco in grado di “allungare” i bambini che crescono poco si ripresenta ciclicamente. Il GH dilaga dagli USA all’Europa, e l’occasione per rilanciarne l’uso è un allargamento delle possibilità di prescrizione che c’è stato negli Stati Uniti e che potrebbe estendersi anche alla nostra Europa. Finora, infatti, questa sostanza era consigliata come terapia per alcune patologie specifiche.
Indicazioni e Controversie:L’ormone della crescita ha un sicuro effetto in casi specifici in cui si riesce a dimostrare la carenza dell’ormone, come, per esempio, nell’ipopituitarismo e in poche altre patologie. Tra queste vi sono la sindrome di Turner, la sindrome di Prader-Willi, e l’insufficienza renale cronica. La sindrome di Turner, uno dei disturbi più comuni della crescita, riguarda le femmine ed è una sindrome genetica causata dall’assenza o dall’eccesso di cromosomi X. L’incidenza è di 1 ogni 2.500 bambine nate vive. Fu descritta per la prima volta dal Dr. Henry Turner nel 1938 e alcuni tra i sintomi evidenti sono la bassa statura, collo corto o palmato, infantilismo sessuale. Evidenze indicano che l’inizio della terapia sostitutiva con estrogeni all’età di dodici anni permetta una normale progressione puberale senza interferire negativamente sull’efficacia della terapia sostitutiva con ormone della crescita. L'altezza SDS iniziale e la dose di GH (sia iniziale sia durante la pubertà) non sono risultate differenti nel gruppo di pazienti con ST e pubertà spontanea e nel gruppo di pazienti con ST e pubertà indotta. Il maggior guadagno staturale indotto dalla terapia con GH in pazienti con ST sia ottenuto prima dell’inizio della pubertà e non si modifichi in base al tipo di terapia estrogenica proposta (transdermica vs. orale).
A queste disfunzioni potrebbe ora aggiungersi il ritardo di accrescimento in utero per cui nascono “corti”, a gestazione completa, il 3-5% dei neonati, destinati generalmente a recuperare nel tempo. La corsa alla prestanza, insieme alla corsa a qualche profitto in più, tende quindi ad allargare l’area dei consumi del GH verso nuove applicazioni. L’ormone della crescita inoltre può essere utilizzato per aumentare la statura dei bambini bassi che però hanno normale funzionamento dell’ipofisi, ma l’uso in tali casi è controverso. Alcuni genitori considerano la bassa statura una sorta di disturbo, mentre molti medici non approvano l’uso dell’ormone della crescita in questi bambini. Per gli altri bambini con crescita limitata non abbiamo purtroppo dati definitivi per affermare se la somministrazione di questo ormone porti a un guadagno di altezza o no. L’unico studio condotto a lungo termine su bambini di bassa statura trattati dopo gli otto anni di età e seguiti fino alla statura finale è stato condotto in Francia e pubblicato recentemente. Mostra un aumento medio di tre centimetri e mezzo. Il che significa che qualcuno ne avrà guadagnati anche quattro, ma che altri non ne hanno conquistati che uno e mezzo, due o nulla.
Rischi e Considerazioni Etiche:Il problema vero è che il trattamento con GH non comporta solo questi oneri; come tutti i trattamenti ormonali, mette in moto l’intero organismo e la sua influenza, specialmente nella fase della crescita, può portare ad alterazioni che potrebbero manifestarsi in età adulta. Boscherini osserva che la questione più grave sta nel rischio di sviluppare nel tempo malattie metaboliche anche gravi, come il diabete mellito tipo adulto. È vero che l’inizio del trattamento con GH provoca un aumento della crescita. Ma i pericoli del domani sembrano spaventare poco rispetto ai desideri di oggi e si stima che i bambini trattati con l’ormone della crescita siano, in Italia, molti di più di quelli che dovrebbero essere. Segno che ci sono medici di manica larga, pronti a dare il GH a chiunque lo chieda, e medici attenti e disposti a negarlo, quando è giusto, anche a costo di essere antipatici agli occhi dei genitori più insistenti.
Indipendentemente dalla causa associata alla bassa statura, l’ormone della crescita è efficace solo se somministrato prima che le ossa smettano di crescere. In Italia la spesa per acquistare questo ormone è tra le prime venti voci del bilancio per l’acquisto di farmaci a carico del Servizio Sanitario Nazionale. Una spesa sproporzionata al numero di casi in cui l’uso del farmaco è ragionevole, che non si giustifica neppure considerando l’abuso che se ne fa come doping fra gli atleti. C’è molto da capire e da fare ancora, tanto da chiedersi se tutto questo lavoro sia così giustificato per affrontare una questione che non sempre assume le caratteristiche di una vera patologia. Dobbiamo, insomma, considerare la bassa statura come una sorta di malattia da trattare con medicinali? È importante affrontare la questione con la consapevolezza dei vantaggi e dei rischi che si corrono. Tutto, o quasi, comincia in una piccola ghiandola posta alla base del cervello, che si chiama ipofisi e che produce l’ormone della crescita. Se questa non funziona e sintetizza un GH scarso o poco attivo il destino di essere basso è quasi certo. Ma ci sono tanti tipi di “bambini piccoli” e le ragioni di una bassa statura possono essere diverse. A cominciare da quella che tutti vediamo e individuiamo subito: essere figli di genitori non alti. La statura di ogni individuo è già scritta nei geni trasmessi da mamma e papà. Ma è come una molla: potrà estendersi al massimo o rimanere compressa secondo l’ambiente in cui si troverà quel bambino. Mai, comunque, potrà allungarsi più di quello che è all’inizio. Un principio che funziona non solo nei singoli individui, ma anche a livello di popolazione, come accaduto nell’ultimo secolo in Italia: tutti più alti di ben dieci centimetri in media, come si può facilmente controllare anche in famiglia, con i nipoti quasi sempre più alti di genitori e nonni. Questo vuol dire che il traguardo di essere alti non ha una posizione fissa.
Il Supporto alle Famiglie: Il Progetto GrowingUp
Il percorso di diagnosi e cura dei disordini della crescita, inclusi il deficit di GH e il panipopituitarismo, è un percorso impegnativo e lungo - dal momento della diagnosi fino al termine della crescita - e coinvolge tutta la famiglia nella vita quotidiana. Come dice Luca Borella, “È una situazione curabile con un adeguato trattamento medico, ma si tratta di un percorso impegnativo e lungo”. In questo contesto, il progetto GrowingUp si pone come risorsa fondamentale, fornendo informazioni chiare, complete e aggiornate, oltre a consigli pratici, per accompagnare le famiglie in questa difficile ma gestibile sfida.
tags: #panipopituitarismo #nel #lattante #farmaci