L'osteogenesi imperfetta, conosciuta anche come la "malattia delle ossa fragili," è una patologia ereditaria che provoca deformità ossee e le rende estremamente fragili. Questo disturbo è causato da mutazioni in alcuni geni e rientra nel gruppo delle osteodisplasie, patologie che disturbano la crescita ossea. L’osteogenesi imperfetta è l’osteodisplasia più conosciuta e si manifesta clinicamente con fragilità ossea. A causa di mutazioni nei geni che rivestono un ruolo importante nello sviluppo del collagene, la maggior parte dei soggetti colpiti presenta una compromissione della sintesi del collagene, uno dei normali componenti delle ossa. Le ossa diventano pertanto fragili e si rompono (fratturano) facilmente. La triade clinica classica dell’OI comprende fragilità ossea, fratture multiple, spontanee o per traumi lievi, sclere blu e sordità precoce. La malattia colpisce maschi e femmine con un rapporto 1:1 e con un’incidenza di 1/20-50.000 nati vivi.
Cos'è l'Osteogenesi Imperfetta? Definizione e Basi Genetiche
L'osteogenesi imperfetta è una malattia genetica rara caratterizzata da una riduzione della resistenza delle ossa, causata da un'alterazione del tessuto connettivo, in particolare della fibra dell’osso, che le rende più suscettibili alle fratture. Le forme più frequenti di osteogenesi imperfetta sono legate a mutazioni dei geni COL1A1 e COL1A2. Questi geni contengono le informazioni per sintetizzare due catene del collagene, la catena alfa1 e la catena alfa2, che rappresentano la principale molecola del tessuto connettivo, il tessuto di collegamento della struttura dell’osso. La mutazione può essere sporadica, il che significa che non è presente e quindi ereditata dai genitori, ma compare 'de novo' nel bambino. Una mutazione de novo implica che la mutazione è avvenuta durante la formazione della cellula uovo o dello spermatozoo oppure nelle primissime fasi di sviluppo embrionale, per quel bambino soltanto, e pertanto nessun altro membro della famiglia sarà malato.
Nel corso degli anni, sono state identificate mutazioni di altri geni, che caratterizzano forme più frequenti in caso di consanguineità dei genitori. Queste forme si manifestano solo se sono presenti due geni mutati, trasmessi da entrambi i genitori, i quali sono portatori sani della mutazione. Questa mutazione è ‘recessiva’, il che significa che la malattia si verifica soltanto se sono mutati entrambi i geni, sia quello ereditato dal padre sia quello ereditato dalla madre. Attualmente, sono circa 20 i geni malattia noti che, se mutati, possono determinare le varie forme di osteogenesi imperfetta.
Sintomi Generali e Manifestazioni Cliniche
I sintomi tipici dell'osteogenesi imperfetta includono ossa deboli che si rompono facilmente. L’osteogenesi imperfetta può variare da lieve a grave. La maggior parte delle persone che ne soffrono presenta ossa fragili e circa il 70% può manifestare perdita dell’udito. In alcuni soggetti, l’osteogenesi imperfetta può far diventare blu la parte bianca dell’occhio, nota come sclera. Questo fenomeno accade perché il colore delle vene traspare dalle sclere anomale, troppo sottili. Esse sono più sottili del normale in quanto il collagene non si è formato in modo corretto.
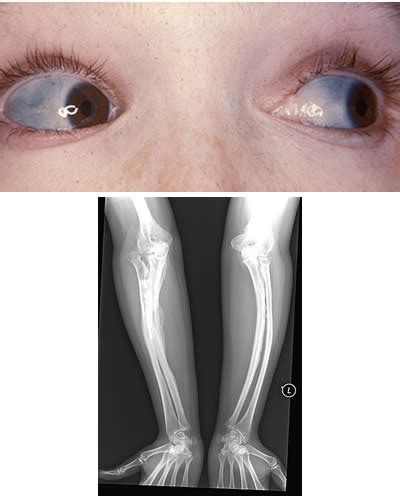
I bambini possono presentare denti poco sviluppati e dal colore anomalo, una condizione chiamata dentinogenesi imperfetta, a seconda del tipo di osteogenesi imperfetta. I denti fragili e trasparenti sono una caratteristica di questa condizione. Possono inoltre associarsi deformità ossee, ecchimosi, lassità ligamentosa e bassa statura. Un'altra manifestazione comune è l'iperlassità delle articolazioni, che si traduce in una riduzione abnorme della tensione dei legamenti. Possono anche verificarsi alterazioni dell'udito, soprattutto tra la seconda e la quarta decade di vita. In alcuni casi, i bambini con osteogenesi imperfetta possono sviluppare patologie cardiache o polmonari. Il dolore muscoloarticolare provocato dalla lassità articolare è anch'esso un sintomo riscontrabile.
I sintomi e i segni dell'osteogenesi imperfetta sono presenti in modo variabile e con diversa gravità, determinando evoluzioni e prognosi molto differenti da caso a caso. Possono essere presenti anomalie craniche e, nelle forme più severe, si possono riscontrare deformità del torace, quali pectus carenatum o pectus excavatum, e un torace a botte. La compressione vertebrale e la scoliosi sono altre manifestazioni scheletriche. È anche possibile un ritardo nello sviluppo neuro-motorio.
La Classificazione dell'Osteogenesi Imperfetta: Dai Tipi Principali alle Varianti Rare
Esistono 5 tipi principali di osteogenesi imperfetta (I, II, III, IV e V), oltre a vari altri tipi più rari, causati da mutazioni in geni diversi. Storicamente, sono stati distinti quattro tipi principali (I-IV), ma la ricerca genetica ha portato all'identificazione di ulteriori varianti, arrivando fino al tipo XVIII, ciascuna definita in base all'alterazione genica specifica.
Osteogenesi Imperfetta Tipo I
L’osteogenesi imperfetta di tipo I è la forma più leggera, spesso definita non deformante con sclere blu. I bambini con questo tipo di osteogenesi imperfetta possono presentare un rischio aumentato di fratture durante l’infanzia, le quali iniziano con la deambulazione e si riducono dopo la pubertà. Alcuni bambini presentano solamente i sintomi delle sclere blu e del dolore muscoloarticolare provocato dalla lassità articolare. La statura è normale o lievemente ridotta, e possono essere presenti cifosi e scoliosi con iperestensibilità articolare. In certi casi, si associa a dentinogenesi imperfetta. Le mutazioni associate a questo tipo sono spesso premature stop codon in COL1A1. È considerata la forma clinicamente meno grave e esordisce nell’infanzia con la triade sintomatologica classica.
Osteogenesi Imperfetta Tipo II
L’osteogenesi imperfetta di tipo II è la forma più grave ed è letale, anche conosciuta come forma congenita o letale perinatale o Sindrome di Vrolik. La fragilità ossea è accentuatissima con fratture multiple che si manifestano quando il feto è ancora in utero, e fratture multiple di coste e ossa lunghe alla nascita. I bambini con OI grave di solito nascono con molte ossa rotte. Il cranio può essere così soffice da non proteggere il cervello dalla pressione applicata alla testa durante il parto. Ciò può provocare emorragie nel cervello o attorno ad esso e morte prima della nascita. Questi bambini hanno braccia e gambe corte, deformità gravi, ossa lunghe tozze e allargate, minima mineralizzazione cranica e le sclere scure. Il neonato affetto da questo tipo può morire improvvisamente nei primi giorni o nelle prime settimane di vita. La OI tipo II porta a morte entro il primo anno di vita. Le mutazioni associate sono solitamente la sostituzione di una Glicina in COL1A1 o COL1A2.
Osteogenesi Imperfetta Tipo III
L’osteogenesi imperfetta di tipo III è il tipo non letale più grave, una forma progressivamente deformante. I bambini affetti da questo tipo di OI sono molto bassi, hanno la colonna vertebrale curva e incorrono in frequenti fratture. Questa forma fa sì che le ossa si rompano frequentemente a seguito di incidenti banali, solitamente quando il bambino inizia a camminare. I bambini affetti da questa malattia sono macrocefali e hanno un viso triangolare a causa del sovrasviluppo della testa e del sottosviluppo delle ossa facciali. Le deformità del torace sono frequenti, mentre il colore delle sclere è variabile, spesso grigiastre. La severa scoliosi è comune, e la dentinogenesi imperfetta è presente. Le mutazioni associate sono spesso la sostituzione di una Glicina in COL1A1 o COL1A2. Sebbene permetta la sopravvivenza, causa deformazioni ossee progressive spesso irreversibili.
Osteogenesi Imperfetta Tipo IV
L’osteogenesi imperfetta di tipo IV varia ampiamente in gravità ed è considerata una forma comune moderata con sclere normali o biancastre. I bambini con questo tipo di OI hanno le ossa che si fratturano facilmente durante l’infanzia e prima della pubertà. Generalmente le sclere sono bianche. Il bambino è di bassa statura, ma la statura è normale o poco ridotta. La fragilità ossea è lieve o moderata, con fratture postnatali e deformità variabili. L'udito è solitamente normale. In certi casi, si associa a dentinogenesi imperfetta. Le mutazioni associate sono anch'esse spesso la sostituzione di una Glicina in COL1A1 o COL1A2. I bambini con questo tipo di OI possono ottenere benefici dal trattamento e il tasso di sopravvivenza è elevato. Questa forma è generalmente compatibile con una normale aspettativa di vita.
Osteogenesi Imperfetta Tipo V
L’osteogenesi imperfetta di tipo V è una forma di grado moderato. Può includere l’indurimento della membrana tra le ossa dell’avambraccio, che limita la mobilità. I bambini presentano lussazione di un osso del braccio (radio) a livello del gomito. Le ossa possono crescere in modo anomalo quando guariscono dalle fratture. La statura è da moderatamente a leggermente bassa, e le sclere sono bianche. A livello microscopico, sono presenti alterazioni caratteristiche del tessuto osseo. È legato a mutazioni del gene IFIMT5.
Altri Tipi di OI (VI-XVIII)
Sono stati identificati altri tipi di osteogenesi, come i tipi VII e VIII, associati rispettivamente a mutazioni dei geni CRTAP e LEPRE1. Più recentemente, altri tipi (6-18) sono stati definiti sulla base dell’alterazione genica specifica e dell'aspetto istologico del tessuto osseo (per esempio i tipi V e VI).
Diagnosi dell'Osteogenesi Imperfetta: Dalla Valutazione Prenatale a Quella Postnatale
La diagnosi di osteogenesi imperfetta si basa principalmente sulla valutazione clinica e sull'analisi di vari indicatori.
Diagnosi Prenatale
Prima della nascita, l’osteogenesi imperfetta può essere diagnosticata nelle gestanti mediante un’ecografia prenatale. Con l’ecografia si possono rilevare deformità, fratture, dimensioni ridotte o altre anomalie ossee. Per formulare la diagnosi, i medici possono eseguire altri esami, come un prelievo dei villi coriali o un’amniocentesi. I test genetici prenatali possono rilevare tutte le forme di OI se le alterazioni genetiche in causa sono già note. Si tratta in questo caso in genere di casi familiari. In caso di una diagnostica prenatale senza che si conosca la mutazione, bisogna affidarsi ad un laboratorio abilitato alla diagnostica prenatale i cui tempi di risposta siano molto rapidi.
Studio dell'addome fetale
Diagnosi Postnatale e Percorso Diagnostico Terapeutico
Dopo la nascita, la diagnosi di osteogenesi imperfetta si basa sui sintomi e sui risultati di un esame obiettivo. Il percorso diagnostico terapeutico include un'accurata anamnesi, che comprende la storia familiare, personale, l'eventuale diagnosi prenatale e la presenza di fratture alla nascita. All'esame obiettivo, il medico ricerca segni specifici come le sclere blu, la dentinogenesi imperfetta, il pectus carenatum o pectus excavatum, il torace a botte, la compressione vertebrale, la scoliosi, la lunghezza degli arti superiore o inferiore alla norma, anomalie craniche, la perdita uditiva e il ritardo nello sviluppo neuro-motorio. Anche un questionario alimentare può fornire informazioni utili.
Se la diagnosi non è chiara, il medico può prelevare un campione di pelle per l’esame al microscopio (biopsia) e analizzare un tipo di cellule del tessuto connettivo chiamate fibroblasti, oppure può prelevare un campione di sangue per analizzare determinati geni. Le radiografie possono mostrare anomalie delle strutture ossee che suggeriscono la presenza di osteogenesi imperfetta. Il quadro radiologico è quasi sempre indicativo, ma varia a seconda dell’età e della forma clinica. In epoca neonatale, è necessaria una valutazione radiologica completa che mette in evidenza le fratture, le deformità dei segmenti ossei e la mineralizzazione incompleta del cranio. In epoca successiva (fino ai 4-5 anni), nelle forme moderate, è tipica una modalità di ossificazione del cranio a “zolle” (ossa wormiane).
Gli accertamenti ematochimici includono emocromo+FL, calcemia, fosforemia, calciuria, fosfaturia, creatinina, proteine totali+ protidogramma, 25OHvitD, 1,25(OH)2vitD, fosfatasi alcalina ossea, desossipiridinoline urinarie, PTH, GH, IGF-1, FSH, LH, TSH. I neonati con OI presentano livelli di fosfatasi alcalina normali o elevati nel sangue periferico.
Gli accertamenti strumentali comprendono RX quando necessario, MOC DXA ogni due anni, QUS e pQCT.
Durante tutta l’infanzia viene eseguito spesso un esame audiometrico per monitorare l’udito, con una valutazione specialistica ogni due anni, poiché la riduzione dell’udito si verifica nel 30% dei pazienti.
La consulenza genetica è fondamentale. Se la valutazione clinica indica la possibilità di OI, si esegue il test genetico molecolare sul potenziale paziente. Fino ad alcuni anni fa, veniva eseguito prima un test per le forme dominanti di OI (geni COL1A1 e COL1A2), e solo successivamente i test per le forme recessive di OI. Attualmente, con le metodiche NGS (Next Generation Sequencing), si studia il pannello completo dei geni di tutte le forme in una unica seduta. Questa analisi genetica molecolare (che conferma la diagnosi clino-radiologica ma non la sostituisce) è in grado di analizzare in una sola tappa tutti i geni le cui mutazioni determinano le varie forme di osteogenesi imperfetta. Una diagnosi molecolare completa è fondamentale per differenziare le varie forme, che possono (e potranno ancora di più in futuro) trovare tipi di cura specifici a seconda dei geni mutati. Vista l’ereditarietà della malattia, in presenza di un caso indice, è indicato lo screening dei familiari. Tuttavia, vi possono essere casi di mutazioni geniche di nuova insorgenza, senza che vi siano familiari affetti.
Valutazioni Specialistiche
Oltre all'audiologica, sono necessarie valutazioni cardiologiche, respiratorie (ogni due anni, per monitorare prolasso aortico e/o mitrale, insufficienza respiratoria per debolezza muscolare, scoliosi importante, fratture costali), oculistiche (ogni due anni, per le caratteristiche sclere blu e la miopia), odontoiatriche (ogni anno a seconda della presenza della dentinogenesi imperfetta o cisti ossee radiolucenti), fisiatriche (soprattutto nelle forme più severe) e ortopediche (in caso di intervento o biopsia ossea).
Diagnosi Differenziale
Non sempre, in caso di fratture ricorrenti, siamo in presenza di OI. Altre patologie caratterizzate da fragilità ossea e difettosa mineralizzazione possono essere in età neonatale l’ipofosfatasia o gravi stati di carenza materno-fetale di vitamina D o l’osteopenia del prematuro (Metabolic Bone Disease of Prematurity). Nelle epoche successive, si devono considerare l’osteomalacia, l’osteopetrosi o difetti dell’osso come displasia fibrosa e tumori. È importante sottolineare che l’abuso su minore è una causa frequente di fratture multiple, con massima incidenza nel primo anno di vita. Differenziare clinicamente le forme di abuso da quelle di OI lieve-moderata può non essere semplice, soprattutto in assenza di una familiarità nota per OI. Le indagini strumentali (TC, densitometria ossea) sono un utile supporto alla diagnosi differenziale, ma la valutazione clinica resta fondamentale.
Gestione e Trattamento dell'Osteogenesi Imperfetta
Non esiste una cura per l’osteogenesi imperfetta, ma sono disponibili trattamenti per gestire i sintomi e alcune complicanze. Il trattamento dell’osteogenesi imperfetta deve avere un approccio multidisciplinare: medico, ortopedico, fisioterapico e riabilitativo. Importante è la prevenzione delle deformità e la limitazione delle disabilità motorie, oltre ad un adeguato sostegno psicologico.
Terapia Farmacologica
Un tipo di farmaco chiamato bifosfonato può aiutare a rafforzare le ossa e ridurre il dolore e il rischio di fratture e la curvatura della colonna vertebrale. I bifosfonati possono essere somministrati per via endovenosa (per esempio pamidronato o acido zoledronico) o per via orale (alendronato). In Italia, il bifosfonato con indicazione per OI è il Neridronato e.v. Diversi studi hanno dimostrato l’utilità dei bifosfonati, sia in formulazione endovenosa che orale, nell’aumentare la massa ossea, riducendo il rischio di frattura, in alcuni pazienti con OI. La somministrazione di questi farmaci deve comunque avvenire previa valutazione e indicazione dello specialista e sotto controllo clinico, in quanto non sono ancora del tutto noti gli effetti di tali farmaci sui bambini nel lungo termine, in particolare sulla crescita ossea.
Le iniezioni di ormone della crescita, in aggiunta ai bifosfonati, possono migliorare la crescita e la forza ossea in alcuni bambini. Alcuni pazienti beneficiano di trattamenti farmacologici con ormone della crescita. Il denosumab è un farmaco simile ai bifosfonati che aiuta a prevenire la perdita ossea e viene normalmente somministrato mediante iniezione. Può aiutare alcuni soggetti affetti da osteogenesi imperfetta. La teriparatide è una forma sintetica di ormone paratiroideo. Questo farmaco stimola la formazione ossea e aumenta la forza. Viene somministrato come iniezione sotto la pelle, ma la teriparatide non può essere somministrata ai bambini.
La vitamina D è una vitamina che aiuta l’organismo ad assorbire calcio e fosforo, essenziali per avere ossa sane. Se i soggetti affetti da osteogenesi imperfetta non possiedono una quantità sufficiente di vitamina D (carenza di vitamina D), i medici prescrivono integratori di vitamina D.

Trattamenti Ortopedici e Riabilitativi
Il trattamento delle ossa fratturate è identico a quello dei bambini che non presentano osteogenesi imperfetta. Tuttavia, le fratture ossee nei bambini affetti da osteogenesi imperfetta possono deformarsi o non crescere affatto. Di conseguenza, nei bambini con fratture multiple, l’accrescimento corporeo può essere permanentemente rallentato e le deformità sono comuni. Per stabilizzare la crescita e prevenire le fratture, i medici possono impiantare chirurgicamente barre metalliche nelle ossa lunghe come le braccia e le gambe. I bambini con OI sono sottoposti a multipli interventi ortopedici correttivi e riparativi delle loro fratture.
La fisioterapia e la terapia occupazionale aiutano a migliorare la funzione e la forza muscolare. I bambini con OI sono avviati a cicli di fisioterapia riabilitativa o all’utilizzo di supporti fisici. Al fine di prevenire le fratture, è importante prendere precauzioni che evitino anche i minimi traumi. Alcuni bambini con perdita dell’udito possono essere aiutati mediante un impianto cocleare, un dispositivo che converte le onde sonore in segnali elettrici inviati a elettrodi impiantati nell’orecchio interno.
Risultati promettenti, ma tuttora oggetto di studio, arrivano da alcuni lavori sul trapianto di midollo osseo da donatore sano per pazienti con OI.
Aspetti Specifici e Ricerca Recente sull'OI Fetale e Neonatale
In epoca neonatale, nelle forme gravi, possono essere presenti fratture e alterazioni degli arti e del cranio che orientano verso una OI già al momento della visita neonatale. Nelle forme moderate o in alcune forme recessive (es. tipo VI), le fratture non sono presenti in epoca neonatale ma iniziano quando il bambino comincia a camminare. Le fratture che si verificano a causa di un trauma minimo o nullo sono spesso la prima indicazione che un neonato o un bambino può avere l’OI. I bambini con forme moderate o gravi di OI nascono spesso con fratture. I bambini con OI più lieve (Tipo I) spesso subiscono la prima frattura svolgendo una normale attività (per esempio durante il cambio del pannolino, mentre vengono sollevati o quando iniziano a stare in piedi e camminare).
L’OI rimane principalmente una diagnosi clinica. Un medico che abbia familiarità con tutti i tipi di OI, può spesso diagnosticare la patologia non solo per la presenza di fratture ma anche sulla base di altre caratteristiche cliniche. Una storia familiare per la malattia e/o test genetici possono confermare una diagnosi.
Uno studio condotto da un gruppo di ricercatori statunitensi ha analizzato sistematicamente la più grande coorte di individui affetti (540 soggetti) arruolati fino ad oggi nei centri di ricerca clinica, e i risultati sono stati pubblicati sulla rivista Genetics in Medicine. I tassi di frattura alla nascita, auto-riferiti, sono stati confrontati tra gli individui con osteogenesi imperfetta di tipo I, III e IV. Come previsto, gli individui con il tipo III hanno avuto il più alto tasso di fratture alla nascita (92,6%), seguiti da quelli con il tipo IV (50,7%) e il tipo I (17,2%).
È emerso che i tassi di fratture non erano diversi a seconda se il parto fosse avvenuto per via vaginale o per taglio cesareo. Solo un maggiore peso alla nascita è risultato collegato a un più alto rischio di fratture, indipendentemente dal tipo di parto. La macrosomia fetale, in generale, è stata associata ad un aumento dei traumi durante il parto e i feti macrosomici (con un peso superiore ai 4 kg) hanno avuto tassi più alti di frattura clavicolare e della spalla. La frattura in utero, la storia materna di osteogenesi imperfetta e la presentazione podalica erano forti predittori per la scelta del taglio cesareo.
Questo studio, il più grande ad aver analizzato l’effetto di vari fattori sul tasso di fratture alla nascita in questa patologia, mostra che il taglio cesareo non è associato a una diminuzione dei tassi di fratture. Con la limitazione che in questa coorte i dati sono stati auto-riportati, questi risultati suggeriscono che il taglio cesareo dovrebbe essere eseguito solo per altre indicazioni materne o fetali, e non al solo scopo di prevenire le fratture.

L’aspettativa di vita per i pazienti affetti da osteogenesi imperfetta dipende dalla gravità della patologia e dalla tempestività e qualità della presa in carico. La prognosi è molto variabile a seconda della forma di osteogenesi imperfetta. Le fratture possono manifestarsi solo con l'inizio della deambulazione e ridursi dopo la pubertà nella forma di tipo 1. Alcune forme (tipo 3) possono essere gravemente deformanti con disabilità irreversibili.
tags: #osteogenesi #imperfetta #fetale