La leucodistrofia metacromatica (MLD) rappresenta una delle sfide più complesse nell'ambito delle malattie neurometaboliche. Si tratta di una patologia neurodegenerativa progressiva di origine genetica, estremamente rara, che appartiene al vasto gruppo delle malattie da accumulo lisosomiale. Nonostante la sua rarità, la MLD non può essere ignorata dalla comunità clinica, poiché il panorama terapeutico ha subito una trasformazione radicale negli ultimi anni, rendendo la diagnosi precoce, spesso già in epoca prenatale, il vero spartiacque tra una storia naturale infausta e la possibilità di un intervento salvavita.
La natura della patologia: basi biochimiche e genetiche
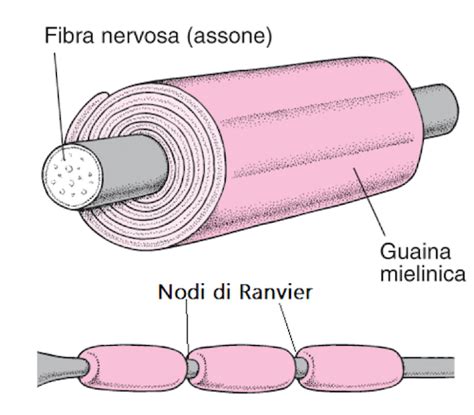
La leucodistrofia metacromatica è causata, nella maggior parte dei casi, dal deficit dell’enzima arilsolfatasi A (ARSA). Questo enzima è fondamentale per il corretto funzionamento dei lisosomi, gli organelli cellulari deputati alla degradazione e al riciclo delle sostanze di scarto del metabolismo. In condizioni di salute, l'ARSA metabolizza il 3-solfato cerebroside, meglio noto come solfatide, un componente essenziale della guaina mielinica che riveste gli assoni nel sistema nervoso centrale e nel sistema nervoso periferico.
Quando l'attività dell'ARSA è deficitaria o assente a causa di mutazioni localizzate sul cromosoma 22, i solfatidi non vengono correttamente smaltiti e si accumulano in modo tossico nelle cellule, in particolare a livello della guaina mielinica. Questa accumulazione determina la demielinizzazione, ovvero la distruzione dei tessuti che avvolgono e isolano i nervi. Poiché la guaina mielinica è indispensabile per la corretta conduzione degli impulsi elettrici, il suo danneggiamento comporta un rallentamento o un blocco dei segnali nervosi, causando movimenti scoordinati, spasticità e una progressiva regressione delle funzioni motorie e cognitive.
In una percentuale molto ristretta di casi, la malattia non è legata direttamente al gene ARSA, ma a mutazioni nel gene PSAP, responsabile della produzione delle saposine, proteine che fungono da attivatori dell'enzima. Anche in questa variante, il risultato finale è l'incapacità di degradare i solfatidi, con conseguenze cliniche sovrapponibili.
Classificazione clinica e sintomatologia
La gravità e l’età di esordio della MLD sono correlate alla quantità di attività enzimatica residua. La letteratura scientifica distingue quattro forme principali, classificate in base al momento della comparsa dei sintomi:
- Forma tardo-infantile: insorge tra i 6 mesi e i 2 anni e mezzo di vita. È la forma più frequente e severa. I primi segnali sono spesso subdoli, come difficoltà nella deambulazione, ipotonia, atrofia ottica e alterazioni della postura. Segue una rapida regressione motoria e cognitiva.
- Forma giovanile precoce: si manifesta tra i 2 anni e mezzo e i 6 anni.
- Forma giovanile tardiva: esordisce in un arco di tempo compreso tra i 6 e i 12 anni.
- Forma adulta: inizia dopo i 12 anni, talvolta oltre i 15-16. L'esordio in questa forma è particolarmente subdolo e spesso caratterizzato da disturbi psichiatrici, declino cognitivo fino alla demenza, atasia e tremore.
Indipendentemente dalla forma, il decorso porta a un deterioramento irreversibile. I bambini perdono la capacità di parlare, sebbene nelle fasi intermedie comunichino ancora attraverso lo sguardo, la risata o il pianto. Nelle fasi terminali, la compromissione della vista e dell'udito si associa alla disfagia, rendendo necessario il supporto nutrizionale tramite gastrostomia.
L’importanza cruciale della diagnosi prenatale
Data la natura progressiva e, fino a poco tempo fa, inarrestabile della patologia, la diagnosi precoce è fondamentale. Nelle famiglie in cui è già presente un caso di MLD, o dove i genitori sono stati identificati come portatori sani del gene mutato, è possibile ricorrere alla diagnosi prenatale.
L’amniocentesi, effettuata solitamente a partire dalla 15ª settimana di gravidanza, permette di prelevare il liquido amniotico per analizzare il corredo genetico del feto. Attraverso tecniche di sequenziamento del DNA, è possibile identificare le mutazioni a carico del gene ARSA prima della nascita. Questa procedura rappresenta un atto di estrema responsabilità clinica: identificare il feto affetto consente di pianificare l'intervento terapeutico immediatamente dopo la nascita, massimizzando le probabilità di successo delle terapie avanzate.
Oltre all'analisi genetica specifica, l'evoluzione tecnologica ha introdotto l'amniocentesi genomica (PrenatalWES), che consente di analizzare l'esoma completo. Sebbene non sia un test diagnostico per ogni singola patologia esistente, esso offre una visione profonda sulle anomalie cromosomiche e sui geni associati a oltre 1200 malattie genetiche note, rappresentando uno strumento potente per la consulenza genetica moderna.
Screening e diagnosi prenatale - Dott. Luigi Caserta
Strumenti diagnostici post-natali
Nel caso in cui la diagnosi non sia avvenuta in epoca prenatale, il medico deve sospettare la MLD sulla base di sintomi neurologici aspecifici. Il percorso diagnostico si avvale di:
- Dosaggio dell'attività enzimatica: la misurazione dell'attività dell'arilsolfatasi A nei leucociti o nei fibroblasti cutanei è un test cardine per confermare il sospetto.
- Analisi biochimica: il dosaggio dei sulfatidi nelle urine (solfatiduria) rivela spesso un accumulo significativo.
- Neuroimaging: la risonanza magnetica cerebrale (RMI) è essenziale per evidenziare le alterazioni tipiche della sostanza bianca, spesso visibili prima della comparsa di segni clinici conclamati.
- Studi di conduzione nervosa: permettono di valutare la velocità con cui gli impulsi viaggiano lungo i nervi periferici, risultando spesso alterata a causa del danno alla guaina mielinica.
La svolta terapeutica: la terapia genica
Per decenni, la MLD è stata considerata incurabile, con una gestione limitata alle sole cure palliative. La svolta è giunta a fine 2020 con l'approvazione in Europa - seguita dall'approvazione negli Stati Uniti - della prima terapia genica al mondo, nota come Libmeldy.
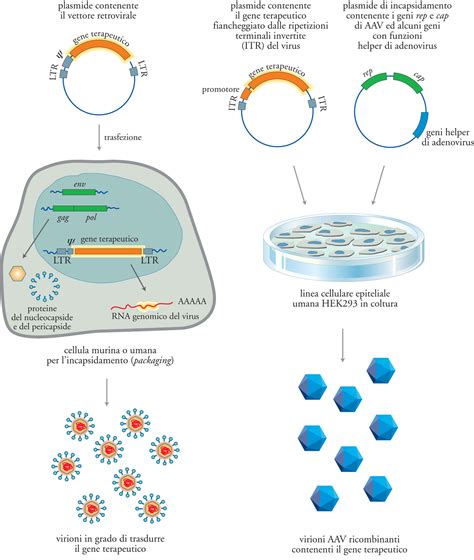
Questo trattamento innovativo rappresenta il coronamento di oltre quindici anni di ricerca condotta dall'Istituto San Raffaele-Telethon per la Terapia Genica (SR-Tiget). Il procedimento prevede il prelievo delle cellule staminali ematopoietiche dal paziente. Queste cellule vengono modificate in laboratorio tramite un vettore lentivirale, derivato da una forma innocua del virus HIV, che inserisce nel DNA delle cellule staminali copie funzionanti del gene ARSA. Una volta reinfuse nel paziente, queste cellule staminali corrette non solo producono l'enzima sano, ma hanno la capacità di attraversare la barriera emato-encefalica, raggiungendo il sistema nervoso centrale. Qui, l'enzima prodotto in sovrannumero viene secreto e assorbito dalle cellule cerebrali circostanti che presentano ancora il gene mutato, correggendo di fatto il deficit biochimico in modo continuativo.
L'efficacia di questa terapia è strettamente legata alla precocità della somministrazione: è indicata principalmente per i bambini con forme tardo-infantile o giovanile-precoce che non abbiano ancora manifestato sintomi gravi o che si trovino in una fase in cui le funzioni motorie e cognitive sono ancora preservate.
Verso lo screening neonatale universale
La disponibilità di una cura capace di modificare la storia naturale della malattia rende il dibattito sullo screening neonatale più acceso che mai. Attualmente, la MLD non è inclusa nello screening neonatale esteso obbligatorio a livello nazionale (Legge 167), ma sono attivi studi pilota in alcune regioni, come la Lombardia.
Il prelievo dal tallone nelle prime ore di vita è l'unica strategia in grado di individuare la malattia in una fase pre-sintomatica, garantendo ai neonati colpiti l'accesso tempestivo alla terapia genica. L'esperienza di associazioni e famiglie, segnate dalla consapevolezza che una diagnosi tardiva preclude l'accesso a cure salvavita, sta spingendo le istituzioni verso l'adozione di protocolli di screening sempre più capillari. L'obiettivo finale rimane l'universalità del test, affinché nessun bambino nasca con una patologia trattabile senza avere la possibilità concreta di riceverne la diagnosi e la cura.
La medicina moderna, grazie alla genomica e alle biotecnologie, sta cambiando il destino di una patologia un tempo definita senza speranza. Il percorso verso la diagnosi precoce richiede però la sinergia tra pediatri, centri di riferimento specializzati e una consapevolezza diffusa nel tessuto sociale, affinché il progresso scientifico si traduca costantemente in pratica clinica e benessere per i piccoli pazienti.
tags: #leucodistrofia #metacromatica #amniocentesi