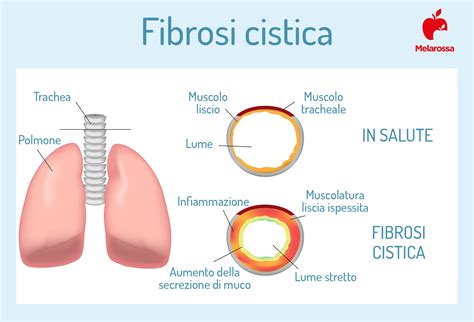
La fibrosi cistica (FC) è una malattia ereditaria che rappresenta la più diffusa patologia genetica grave a livello mondiale. Colpisce circa 1 individuo ogni 2.500-2.700 nati vivi in Italia, con circa 200 nuovi casi diagnosticati ogni anno. Nonostante la sua prevalenza, la fibrosi cistica è una malattia ancora poco nota al grande pubblico, ma di alto interesse sociale, come riconosciuto dalla Legge 23 dicembre 1993, n. 548. Questa condizione multisistemica, che interessa diversi organi, è caratterizzata dalla produzione di muco denso e appiccicoso, anziché fluido e umidificante.
Le Origini della Malattia: Un Difetto Genetico Fondamentale
Alla base della fibrosi cistica vi è un difetto genetico ereditario, trasmesso secondo un meccanismo autosomico recessivo. Ciò implica che per manifestare la malattia, un individuo deve ereditare due copie del gene difettoso, una da ciascun genitore. Il gene in questione è il CFTR (Cystic Fibrosis Transmembrane Conductance Regulator), responsabile della produzione di una proteina cruciale per il corretto trasporto di acqua e sali minerali attraverso le membrane cellulari. Quando questo gene è mutato, la proteina CFTR non funziona in modo appropriato, portando alla produzione di secrezioni corporee, in particolare muco e sudore, con caratteristiche alterate.
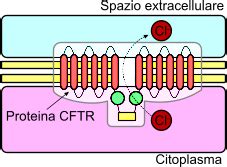
Le mutazioni a carico del gene CFTR sono numerose, con oltre mille varianti identificate, e possono determinare differenti gradi di gravità della malattia. In Italia, la frequenza dei portatori sani del gene CFTR è stimata intorno a 1 su 30, e una coppia su 900 risulta essere composta da due portatori. In tali coppie, il rischio di avere un figlio affetto da fibrosi cistica è del 25% ad ogni gravidanza, mentre vi è una probabilità del 50% di avere un figlio portatore sano e del 25% di avere un figlio non portatore e non affetto. La frequenza dei portatori varia a seconda delle popolazioni, essendo maggiore nelle popolazioni europee e nordamericane.
Il test per l'identificazione del portatore sano è offerto dal Servizio Sanitario Nazionale a persone con parenti affetti da fibrosi cistica o a coppie che intraprendono percorsi di procreazione assistita. Questo test, solitamente effettuato tramite un esame del sangue, può essere proposto in fase pre-concezionale o prenatale. È importante notare che i test di screening non sono perfetti e potrebbero non individuare il 100% dei portatori, poiché non tutte le mutazioni del gene CFTR sono ancora conosciute.
Manifestazioni Cliniche: Un Quadro Multisistemico Complesso
La fibrosi cistica è una patologia multisistemica, il che significa che colpisce diversi organi e apparati del corpo. I sintomi tendono a comparire nella prima infanzia, sebbene in alcuni casi possano manifestarsi subito dopo la nascita o emergere solo in età adulta.
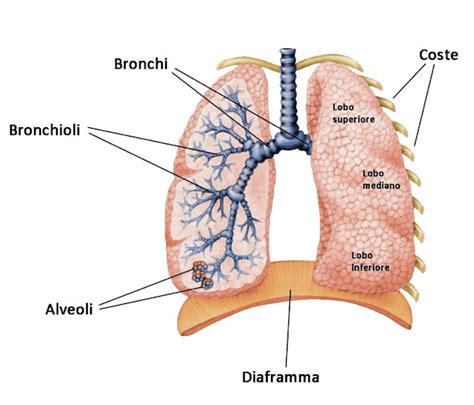
Apparato Respiratorio: Il Cuore dei Problemi
Nei polmoni, il muco denso e viscoso ostruisce le vie aeree, creando un terreno fertile per infezioni batteriche ricorrenti. Queste infezioni, se non trattate adeguatamente, possono cronicizzare, portando a infiammazione, danni progressivi ai tessuti polmonari e, nel tempo, a insufficienza respiratoria. L'ostruzione delle vie aeree può manifestarsi con tosse cronica, produzione di espettorato denso, respiro sibilante e difficoltà respiratorie.
Apparato Digerente: Malassorbimento e Ritardo di Crescita
Il pancreas, uno degli organi più colpiti insieme ai polmoni, vede i suoi dotti ostruiti dal muco. Questo impedisce agli enzimi digestivi di raggiungere l'intestino tenue, compromettendo la digestione e l'assorbimento dei nutrienti, in particolare dei grassi e delle vitamine liposolubili (A, D, E, K). Di conseguenza, i pazienti con fibrosi cistica possono soffrire di diarrea, malassorbimento, ritardo della crescita nei bambini e malnutrizione negli adulti. Il progressivo danneggiamento del pancreas può anche portare, con l'età, allo sviluppo di una forma di diabete.
Intestino Tenue: Ostruzione Neonatale Precoce
Nei neonati, l'ostruzione intestinale è una delle prime manifestazioni cliniche, nota come ileo da meconio. Il meconio, le prime feci del neonato, risulta eccessivamente denso e appiccicoso a causa della fibrosi cistica, causando un blocco intestinale che richiede un intervento medico rapido per prevenire complicanze come la perforazione intestinale e la peritonite.
Fegato e Dotti Biliari: Rischio di Colestasi
Il muco può ostruire anche i dotti biliari, che collegano il fegato e la cistifellea all'intestino tenue. Questo può portare a stasi biliare o colestasi, in cui la bile ristagna nel fegato e può raggiungere il circolo sanguigno. Circa il 25-30% dei soggetti con fibrosi cistica può sviluppare un coinvolgimento epatico, che può manifestarsi con l'ingrossamento del fegato e alterazioni dei valori epatici.
Ghiandole Sudoripare: Sudorazione Eccessiva e Perdita di Sali
Le persone affette da fibrosi cistica tendono a sudare in misura maggiore rispetto alla norma, con una conseguente perdita eccessiva di sali minerali. Questo squilibrio elettrolitico può portare a disidratazione, aumento della frequenza cardiaca, affaticamento, debolezza e riduzione della pressione sanguigna.
Sistema Riproduttivo: Infertilità Maschile e Difficoltà nel Concepimento
Nei maschi, la fibrosi cistica è spesso associata all'infertilità. Si stima che tra il 97% e il 98% degli uomini affetti da questa patologia presentino un'assenza bilaterale congenita dei dotti deferenti. Il muco denso ostruisce questi dotti, impedendo il passaggio degli spermatozoi nel liquido seminale (azoospermia ostruttiva). Anche nelle donne possono verificarsi disturbi alle ovaie, con conseguenti difficoltà nel concepimento.
Diagnosi Precoce: Fondamentale per una Migliore Gestione
La diagnosi precoce della fibrosi cistica è fondamentale per garantire un trattamento tempestivo e migliorare la qualità e l'aspettativa di vita dei pazienti.
Screening Neonatale: Una Finestra Cruciale
Oggi, la maggior parte dei casi di fibrosi cistica viene diagnosticata subito dopo la nascita attraverso un esame di controllo obbligatorio per legge, lo screening neonatale. Questo test consiste nel prelevare una piccola goccia di sangue dal tallone del neonato al secondo-terzo giorno di vita e misurare i livelli di una sostanza, la tripsina immunoreattiva. Livelli elevati di tripsina immunoreattiva possono indicare la presenza della malattia e richiedono ulteriori accertamenti.
Screening Neonatale - Intervista alla dott.ssa Spina
Analisi Genetica: Conferma Diagnostica e Identificazione delle Mutazioni
L'analisi genetica rappresenta il "gold standard" per la diagnosi della fibrosi cistica. Questo esame, eseguito su un campione di sangue, saliva o cellule prelevate dalla mucosa orale, consente di identificare le mutazioni nel gene CFTR e confermare la diagnosi, anche nei casi dubbi o nelle forme atipiche. Test avanzati possono rilevare anche mutazioni rare, contribuendo a una comprensione più completa del quadro genetico del paziente. L'analisi genetica riveste un ruolo importante anche nello screening familiare, permettendo di identificare i portatori sani e fornire consulenza genetica e supporto per la pianificazione riproduttiva.
Forme Atipiche o Borderline: Sfide Diagnostiche Aggiuntive
Esistono anche forme atipiche o borderline di fibrosi cistica, meno gravi della forma classica, in cui i sintomi possono essere isolati o più lievi. Queste forme possono presentare manifestazioni come malattia polmonare cronica a inizio tardivo e lieve, sinusite cronica, cirrosi biliare, pancreatite cronica o ricorrente, o infertilità. La diagnosi in questi casi può essere più complessa e richiedere un approccio integrato che combini test clinici, test genetici e test del sudore.
Terapie e Trattamenti: Un Percorso in Continua Evoluzione
Attualmente, non esiste una cura definitiva per la fibrosi cistica, ma la ricerca ha compiuto passi da gigante nello sviluppo di terapie e farmaci in grado di contrastare i sintomi, migliorare la qualità della vita e rallentare la progressione della malattia.
Farmaci Modulatori della Proteina CFTR: Nuove Speranze
Negli ultimi anni, sono stati sviluppati farmaci modulatori della proteina CFTR, in grado di correggere o potenziare il funzionamento della proteina difettosa in presenza di specifiche mutazioni genetiche. Questi farmaci hanno dimostrato un'efficacia significativa nel ridurre i sintomi polmonari e altri sintomi correlati alla fibrosi cistica, migliorando la funzione polmonare e la qualità della vita dei pazienti. Tuttavia, questi trattamenti non coprono tutte le mutazioni del gene CFTR e non sono indicati per tutte le fasce d'età, lasciando ancora una percentuale di pazienti esclusi da queste cure innovative.
Terapie di Supporto: Un Approccio Multidisciplinare
Oltre ai farmaci modulatori, le terapie per la fibrosi cistica si concentrano sull'alleviamento dei sintomi e sulla prevenzione delle complicanze. Queste includono:
- Trattamenti farmacologici: Antibiotici per via orale o endovenosa per trattare le infezioni polmonari, aerosol di antibiotici e farmaci per fluidificare le secrezioni respiratorie.
- Fisioterapia respiratoria: Esercizi quotidiani mirati a rimuovere l'eccesso di muco accumulato nei polmoni, insegnati ai pazienti per essere eseguiti regolarmente a domicilio.
- Nutrizione: Una dieta ipercalorica è spesso raccomandata per garantire un adeguato apporto nutrizionale e contrastare il malassorbimento.
- Trapianto polmonare: In casi di insufficienza respiratoria grave e progressiva, il trapianto polmonare rappresenta un'opzione terapeutica.
La gestione ottimale della fibrosi cistica richiede un'assistenza da parte di un team multidisciplinare composto da medici specialisti, infermieri, fisioterapisti, dietologi, assistenti socio-sanitari e farmacisti, operanti all'interno di Centri multiprofessionali dedicati.
Aspettative di Vita e Prospettive Future: Un Miglioramento Costante
Grazie ai continui progressi nelle terapie, nella diagnosi precoce e nell'assistenza, l'aspettativa di vita delle persone con fibrosi cistica è notevolmente aumentata negli ultimi decenni. Se fino a qualche decennio fa era limitata a pochi anni, oggi oltre il 50% della popolazione con fibrosi cistica in Italia è costituita da adulti e l'aspettativa di vita si aggira intorno ai 40 anni, con una tendenza al miglioramento continuo.
L'impegno della ricerca scientifica, supportato da istituti come l'Istituto Mario Negri e da associazioni di volontariato come la Lega Italiana Fibrosi Cistica e la Fondazione per la Ricerca sulla Fibrosi Cistica, continua a essere fondamentale per sviluppare nuove terapie, migliorare la comprensione della malattia e offrire una migliore qualità di vita ai pazienti. Studi come la valutazione di Health Technology Assessment dello screening del portatore di fibrosi cistica mirano a ottimizzare le strategie di prevenzione e diagnosi, valutando l'impatto economico, sociale ed etico delle diverse opzioni.
Nonostante le sfide che la fibrosi cistica presenta, i progressi compiuti offrono un quadro di speranza per il futuro, con la prospettiva di una vita sempre più quasi normale per le persone che convivono con questa complessa malattia genetica.