L'igroma cistico, una malformazione congenita del sistema linfatico, e la sindrome di Turner, una condizione cromosomica che colpisce esclusivamente le femmine, sono due entità cliniche che, sebbene distinte, presentano un legame significativo, soprattutto in epoca prenatale. La comprensione di questa interrelazione è fondamentale per un'adeguata diagnosi, gestione e counseling delle pazienti e delle loro famiglie.
L'Igroma Cistico: Una Finestra sulle Malformazioni Fetali
L'igroma cistico, noto anche come linfangioma cistico, è una patologia che si manifesta durante lo sviluppo embrionale, caratterizzata dalla formazione di cavità ripiene di liquido linfatico. Queste lesioni si localizzano più frequentemente nella regione del collo (circa il 75% dei casi), ma possono interessare anche le ascelle o altre parti del corpo. Dal punto di vista istologico, l'igroma cistico è costituito da ampi spazi linfatici rivestiti da cellule endoteliali, separati da tessuto connettivo. Sebbene sia tecnicamente una neoplasia benigna dei vasi linfatici, la sua importanza clinica in ambito prenatale è considerevole, poiché spesso funge da marcatore per gravi anomalie cromosomiche o sindromi genetiche complesse.
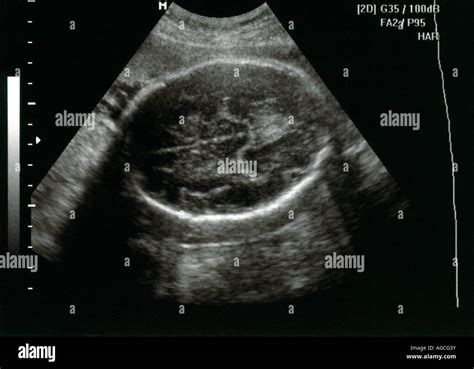
Le cause dell'igroma cistico sono riconducibili a un alterato deflusso del sistema linfatico. Si ipotizza un'alterata linfoangiogenesi o fenomeni ostruttivi che impediscono la normale comunicazione tra i vasi linfatici e il sistema venoso, portando a una dilatazione dei sacchi linfatici laterocervicali. L'igroma cistico può presentarsi in diverse forme: settato, ovvero diviso da membrane interne, o non settato. Le forme settate sono statisticamente più associate ad anomalie del cariotipo.
La diagnosi di igroma cistico viene solitamente posta intorno alla 12ª-14ª settimana di gestazione mediante ecografia ostetrica. L'indagine ecografica permette di valutare l'estensione cranio-caudale e laterale della massa, distinguendo tra forme settate e non settate. Le ecografie seriate, effettuate ogni tre settimane, sono cruciali per monitorare l'evoluzione della lesione e l'eventuale insorgenza di complicanze.
Tra le complicanze più gravi associate all'igroma cistico vi è l'idrope fetale, caratterizzata da un accumulo eccessivo di liquidi in più compartimenti fetali. In casi di igroma cistico di grandi dimensioni, soprattutto se localizzato nel collo, esiste il rischio di ostruzione delle vie respiratorie al momento della nascita, richiedendo un'attenta pianificazione del parto.
Le opzioni terapeutiche per l'igroma cistico in utero includono la scleroterapia, che prevede l'iniezione di sostanze irritanti per indurre il collasso delle cisti, e la chirurgia resettiva per l'asportazione della massa. Tuttavia, la gestione durante la gravidanza è spesso di tipo osservazionale, con un focus sul monitoraggio ecografico.
La Sindrome di Turner: Un'Anomalia Cromosomica con Manifestazioni Diverse
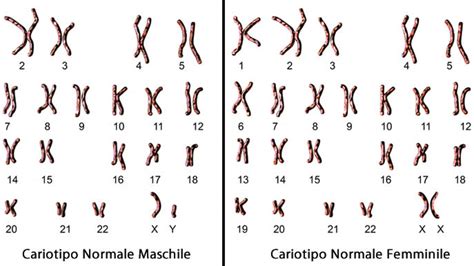
La sindrome di Turner (ST) è una condizione cromosomica che si verifica in circa 1 donna ogni 2.000-2.500 nate vive. È causata dalla presenza di un solo cromosoma sessuale X invece dei due normalmente presenti nel corredo cromosomico femminile (46,XX). L'anomalia può manifestarsi come una monosomia completa (45,X) o come un mosaicismo, in cui alcune cellule presentano un cariotipo normale (46,XX) e altre un solo cromosoma X (45,X). Il mosaicismo, presente in più della metà delle donne affette, può portare a manifestazioni cliniche più sfumate o addirittura assenti.
Le cause della sindrome di Turner sono esclusivamente genetiche e non sono correlate a fattori ereditari, ambientali o a comportamenti dei genitori. L'errore genetico, noto come "non-disgiunzione", avviene durante la formazione degli ovociti o degli spermatozoi.
I segni e i sintomi della sindrome di Turner sono estremamente variabili e possono presentarsi in diverse fasi dello sviluppo, con differente gravità. Tra le caratteristiche cliniche salienti si annoverano:
- Bassa statura: La statura media definitiva delle pazienti non sottoposte a terapia è intorno a 140-142 cm. La crescita staturale rallenta dopo i 4-5 anni, senza lo spurt puberale tipico.
- Infertilità/fertilità sub-normale: Oltre il 90% dei casi è dovuto a disgenesia gonadica, ovvero uno sviluppo mancato o parziale delle ovaie, con conseguente insufficienza ovarica e quasi totale assenza di produzione ormonale. In circa il 10% dei casi di mosaicismo, è possibile una condizione di fertilità.
- Linfedema congenito: Gonfiore di mani e piedi, solitamente regredisce nella prima infanzia, ma può persistere edema sul dorso delle dita.
- Anomalie toraciche: Torace ampio a "scudo", con aumentata distanza dei capezzoli, che possono essere ipoplasici o invertiti.
- Dismorfismi facciali: Palato stretto, mandibola poco sviluppata, epicanto, anomalie auricolari, collo corto, attaccatura posteriore dei capelli bassa e pterigio.
- Anomalie scheletriche: Cubito valgo, anomalie del ginocchio, metacarpo e metatarso corti, displasia ossea.
- Malformazioni renali: Rene a ferro di cavallo, doppio distretto renale e altre alterazioni minori.
- Anomalie cardiache: Valvola aortica bicuspide, coartazione dell'aorta, stenosi della valvola aortica, prolasso della valvola mitrale. Queste anomalie sono presenti nel 15-50% dei casi.
- Ipoacusia: Riduzione dell'udito, in oltre il 50% dei casi, sia trasmissiva che neurotrasmissiva.
- Altre condizioni associate: Obesità, diabete mellito tipo 2, ipertensione, patologie autoimmuni (tiroidite autoimmune, celiachia), ipotiroidismo.
Il quoziente intellettivo nelle bambine e donne con sindrome di Turner è generalmente nella norma, ma possono essere presenti difficoltà di apprendimento, in particolare nelle prestazioni matematiche, nell'organizzazione visuo-spaziale e deficit attentivi. Non va sottovalutato l'aspetto psicologico, poiché la sterilità e le caratteristiche fenotipiche peculiari possono generare ansia, depressione e difficoltà relazionali.
Sindrome di Turner: cos'è, come si diagnostica e come si cura?
L'Associazione tra Igroma Cistico e Sindrome di Turner
L'associazione tra igroma cistico e sindrome di Turner è particolarmente rilevante in epoca prenatale. Circa il 50-60% dei feti con igroma cistico presenta un'anomalia del numero di cromosomi, e la sindrome di Turner (monosomia X) è una delle più frequenti tra queste. L'igroma cistico, soprattutto se non settato, può essere un marker ecografico che indirizza verso la ricerca di anomalie cromosomiche.
In caso di diagnosi ecografica di igroma cistico, è fondamentale un accurato inquadramento diagnostico che include:
- Ecocardiografia fetale: Per la ricerca di cardiopatie funzionali e strutturali, che sono significativamente più frequenti nei feti affetti da igroma cistico rispetto alla popolazione generale.
- Analisi del cariotipo: Ottenibile mediante villocentesi o amniocentesi, per valutare la presenza di aneuploidie. Il rischio di aneuploidia è sei volte maggiore nella popolazione di feti con igroma cistico.
La diagnosi di sindrome di Turner viene confermata attraverso l'analisi del cariotipo. Nei casi di monosomia completa, tutte le cellule presentano l'assetto 45,X, mentre nei soggetti con mosaicismo è presente una quota variabile di cellule 45,X e 46,XX. In alcuni casi, è possibile riscontrare modificazioni strutturali del cromosoma X o la presenza di materiale del cromosoma Y, che espone al rischio di insorgenza di gonadoblastoma.
Management e Follow-up
La gestione della sindrome di Turner è personalizzata e mira a correggere alcuni aspetti del quadro clinico, non esistendo una terapia risolutiva.
- Terapia con Ormone della Crescita (GH): Va iniziata precocemente in caso di rallentamento della crescita o bassa statura. L'utilizzo di GH ad alte dosi e per lunghi periodi può portare a un buon guadagno staturale, con molte pazienti che superano i 150 cm.
- Terapia Ormonale Sostitutiva (TOS) con Estrogeni e Progestinici: Inizia intorno agli 11-12 anni per favorire lo sviluppo puberale e mantenere la salute ossea.
- Preservazione della Fertilità: Nei casi di mosaicismo con attività ovarica residua, è possibile valutare la crioconservazione degli ovuli. Le tecniche di procreazione medicalmente assistita con ovodonazione possono consentire la gravidanza, sebbene queste comportino un monitoraggio multispecialistico e rischi di complicanze cardiovascolari.
- Fisioterapia e Supporto Psicologico: Sono essenziali per affrontare le sfide fisiche e psicologiche legate alla condizione.
Il follow-up della sindrome di Turner deve essere multidisciplinare e a lungo termine, per monitorare la crescita, la funzione ovarica, la salute cardiovascolare, uditiva, metabolica e per fornire supporto psicologico. L'aspettativa di vita per le bambine con sindrome di Turner è oggi buona, sebbene possa essere ridotta in media a causa delle complicanze cardiovascolari.
Conclusioni
L'igroma cistico e la sindrome di Turner rappresentano due importanti sfide diagnostiche e gestionali in ambito ostetrico e pediatrico. La loro associazione sottolinea l'importanza di un approccio integrato che combini l'imaging fetale avanzato, l'analisi genetica e un follow-up clinico attento e personalizzato. La ricerca continua e la collaborazione tra specialisti sono fondamentali per migliorare la comprensione di queste condizioni complesse e offrire alle pazienti le migliori prospettive di salute e benessere.