La sindrome di Noonan (SN) è una malattia multisistemica rara e molto variabile, con un'incidenza stimata tra 1/1000 - 1/2500 nati, sebbene una lieve espressione della condizione possa essere presente in 1/100. Questa condizione è caratterizzata prevalentemente da bassa statura, dismorfismi facciali caratteristici, cardiopatie congenite, cardiomiopatia e un rischio aumentato di sviluppare tumori durante l'infanzia. È una sindrome contraddistinta da bassa statura per femori corti (inferiori al 3° centile), cardiopatia congenita, persistenza della vena ombelicale di destra, edema nucale, igroma cistico, ascite, idrotorace, idrope non immune, anomalie renali, ipertelorismo, orecchie ad impianto basso e ruotate posteriormente. La sindrome è presente sin dalla nascita, ma la diagnosi, essendo ancora basata sulle caratteristiche fenotipiche (complesso dei caratteri individuali morfologici e fisiologici determinato dal patrimonio genetico e dai fattori ambientali), può essere fatta a diverse età. L’età media in cui viene posta la diagnosi è 9 anni.
Origine e Sviluppo dell'Igroma Cistico
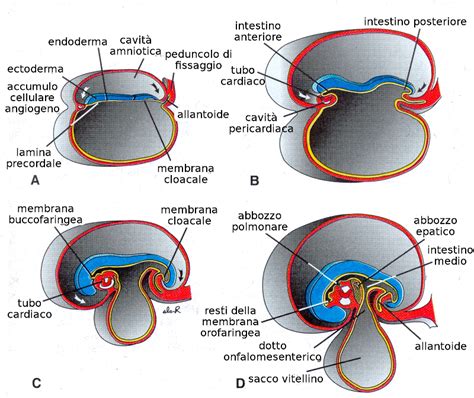
L'igroma cistico, noto anche come linfangioma cistico, rappresenta una rara malformazione congenita del sistema linfatico che insorge durante lo sviluppo embrionale. Questa condizione, caratterizzata dalla formazione di una o più cavità piene di liquido linfatico, è legata a un'alterazione del sistema linfatico durante l'embriogenesi. Si ipotizza che derivi da un'insufficiente drenaggio del sistema linfatico in uno dei suoi punti di connessione con il sistema venoso, in particolare a livello dei sacchi linfatici giugulari. Durante lo sviluppo fetale, il sistema linfatico drena nel sacco linfatico giugulare, una struttura primitiva che, intorno ai 40 giorni di età gestazionale, dovrebbe comunicare con la vena giugulare. Se questa comunicazione si interrompe, a causa di un'alterata linfoangiogenesi o di fenomeni ostruttivi, si verifica una dilatazione dei sacchi linfatici laterocervicali, con conseguente formazione dell'igroma cistico. Dal punto di vista istologico, l'igroma cistico è costituito da ampi spazi linfatici rivestiti da cellule endoteliali, separati da uno stroma di tessuto connettivo. Sebbene tecnicamente sia una neoplasia benigna dei vasi linfatici, la sua rilevanza clinica in ambito prenatale è notevole, poiché spesso funge da marcatore per gravi anomalie cromosomiche o sindromi genetiche complesse.
Caratteristiche Cliniche e Diagnosi Prenatale dell'Igroma Cistico
L'igroma cistico si presenta tipicamente come una tumefazione sottocutanea di consistenza soffice, a decorso generalmente lento. Può essere uni- o multicistico e può interessare diverse parti del corpo, sebbene la localizzazione più frequente sia la regione del collo (circa il 75% dei casi), seguita dalle ascelle. Altre localizzazioni includono il tronco e gli arti. La diagnosi precoce è fondamentale e avviene solitamente nel corso della gravidanza tramite ecografia. L'esame della Translucenza Nucale, eseguito tra la 9ª e la 14ª settimana di gestazione, può rivelare la presenza di questa formazione cistica. Nel primo trimestre possono evidenziarsi NT ispessita, igroma, idrope. In presenza di un igroma cistico, vengono prescritti ulteriori accertamenti per valutare la sua estensione, la presenza di setti interni (forme settate sono statisticamente più correlate ad anomalie del cariotipo) e l'eventuale associazione con altre malformazioni. Le ecografie seriali permettono di monitorare l'evoluzione dell'igroma e l'insorgenza di complicanze come l'idrope fetale, una condizione caratterizzata da un accumulo eccessivo di liquidi in almeno due compartimenti fetali, considerata la più grave complicanza. La biochimica mostra un tri-test positivo.
L'igroma cistico è un quadro malformativo del collo fetale costituito da un’ectasia cistica dei vasi linfatici latero-cervicali e cervico-dorsali dovuto a un alterato deflusso paravenoso dal complesso linfatico latero-cervicale a quello giugulare e da questo al dotto toracico. Per la diagnosi sono utilizzate sia la scansione assiale del collo, che quella sagittale mediana dell’estremo cefalico, possibilmente anche con approccio dorsale: in questo modo sarà possibile valutare l’estensione cranio-caudale e laterale della massa e differenziare l’igroma non settato da quello settato. È patologia che maggiormente sembra associarsi alla diagnosi ecografica di patologia linfatica del collo e che tra i segni ecografici rilevanti e patognomonici associa al rilievo di igroma (soprattutto non settato) la presenza di cardiomiopatia ipertrofica, difetti interatriali e/o interventricolari ma soprattutto stenosi polmonare (che sembra essere il marker complicativo, talvolta letale, del quadro sindromico). Etiologicamente le cause cardiache sono le più frequenti (20-30% dei casi), anche se le cause cromosomiche sono prevalenti nel primo trimestre. La differenziazione principale riguarda la presenza o meno di setti: alcune forme non settate, specialmente se non associate ad aneuploidie né a idrope, possono regredire nel corso della gestazione o alla nascita con un outcome neonatale normale.
MED2000ECO Igroma Cistico
Associazione con Anomalie Cromosomiche e Sindromi Genetiche
Una delle sfide maggiori nella gestione dell'igroma cistico è la sua frequente associazione con anomalie cromosomiche e sindromi genetiche. Si stima che circa il 50-60% dei feti con igroma cistico presenti un'anomalia del numero di cromosomi (aneuploidia). Le anomalie più frequentemente riscontrate sono la sindrome di Turner (45, X) e le tre trisomie autosomiche (trisomia 21, 18 e 13) oltre ad un’ampia gamma di arrangiamenti cromosomici rari (delezioni e traslocazioni sbilanciate). Queste includono:
- Sindrome di Turner (monosomia X): Caratterizzata dall'assenza parziale o totale di un cromosoma X nelle femmine.
- Sindrome di Down (Trisomia 21): Causata dalla presenza di una copia extra del cromosoma 21.
- Sindrome di Edwards (Trisomia 18) e Sindrome di Patau (Trisomia 13): Condizioni più gravi associate a ritardi nello sviluppo e malformazioni multiple.
Inoltre, anche in presenza di un cariotipo normale, l'igroma cistico può essere parte di sindromi genetiche specifiche, tra cui spicca la Sindrome di Noonan. Questa condizione autosomica dominante è associata a difetti cardiaci e caratteristiche facciali distintive. Altre sindromi che possono associarsi all'igroma cistico includono la Sindrome di Fryns, la Sindrome di Neu-Laxova, la Multiple Pterigium Syndrome, la Sindrome di Escobar, la Sindrome di Klippel-Feil e la Sindrome di Zellweger. Le malformazioni associate, che si riscontrano in circa il 60% dei casi, comprendono frequentemente cardiopatie congenite, displasie scheletriche, anomalie del sistema nervoso centrale ed ernia diaframmatica.
La Sindrome di Noonan: Un Dettaglio Approfondito
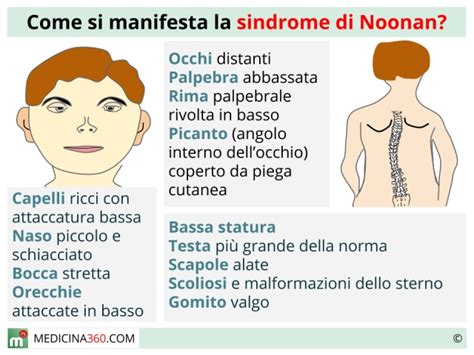
Il fenotipo della Sindrome di Noonan è caratterizzato da bassa statura, ipertelorismo, impianto basso delle orecchie che appaiono anche ruotate posteriormente, collo tozzo con cute ridondante o con pieghe laterali, pieghe epicantiche, sordità, ritardo motorio, difetti della coagulazione per trombocitopenia. I dismorfismi facciali caratteristici sono maggiormente evidenti nell'infanzia: fronte alta e ampia, ipertelorismo, ptosi palpebrale e rime palpebrali oblique verso il basso, orecchie spesse a impianto basso e ruotate posteriormente, filtro profondo, micrognazia, capelli ricci e collo corto, talvolta con pterigio. Con l'età, il viso diventa triangolare, con pliche cutanee marcate. Nel neonato le caratteristiche cliniche sono spesso sfumate. Pertanto se non è presente una cardiopatia congenita o storia familiare positiva; la diagnosi viene posta più tardivamente, di solito nella prima infanzia. Nella prima infanzia è comune il riscontro di problemi alimentari (in particolare difficoltà di suzione e vomito) ed in alcuni casi è stata necessaria la nutrizione mediante sondino naso-gastrico. I primi problemi clinici che si manifestano nella sindrome di Noonan sono la cardiopatia congenita e le difficoltà di alimentazione e di crescita. Le difficoltà di alimentazione migliorano di solito nel tempo.
Cardiopatie Congenite nella Sindrome di Noonan
La cardiopatia congenita è nel 40-50% dei casi rappresentata da stenosi polmonare, o da cardiomiopatia ipertrofica (20-30%), DIV, DIA, Tetralogia di Fallot. La cardiopatia congenita più comune è la stenosi della valvola polmonare (50-60%) con displasia della valvola polmonare e diverse malformazioni cardiache (difetti del setto interatriale e interventricolare, ecc.). È comune la cardiomiopatia ipertrofica ad esordio prenatale (20%), che può essere stabile o a progressione rapida. Le cardiopatie congenite più frequenti sono la stenosi valvolare polmonare e la cardiomiopatia ipertrofica. Possono essere presenti però anche difetti del setto o coartazione aortica. La principale causa di morbidità e mortalità è rappresentata da una grave patologia cardiaca. La stenosi della valvola polmonare è una ristrettezza della valvola che porta il sangue dal cuore ai polmoni per essere ossigenato.
Altre Manifestazioni Cliniche della Sindrome di Noonan
Il 50% dei pazienti presenta ritardo della crescita, raramente associato al deficit dell'ormone della crescita. I pazienti hanno difficoltà a guadagnare peso e molti di essi mantengono una corporatura snella per tutta la vita. I principali segni clinici ortopedici comprendono la deformità dello sterno, i piedi equino-vari e la scoliosi progressiva (che esordisce nell'adolescenza). Spesso la pelle è secca e, talvolta, ipercheratosica sulle mani e sui piedi. I capelli sono ricci e possono essere folti o radi. I pazienti possono presentare linfedema periferico, che in alcuni può essere progressivo ed esteso. Sono comuni le anomalie oculari (strabismo, errori refrattivi) e l'affastellamento dei denti. Il 10% dei pazienti presenta sordità. Nel 30-40% dei pazienti si osserva ritardo nel linguaggio e difficoltà di apprendimento, e nel 10-20% disabilità intellettiva (spesso lieve). Sono comuni la disprassia (goffaggine), il deficit dell'attenzione, l'agitazione, i disturbi dell'umore e quelli emotivi, così come le difficoltà nell'identificare e nell'esprimere le emozioni, che possono portare a difficoltà nelle interazioni sociali. Lo sviluppo motorio e la pubertà sono ritardati e il 50% dei pazienti presenta bassa statura. Due terzi dei maschi presentano criptorchidismo mono- o bilaterale; l'ipofertilità, se presente, interessa solo i maschi. La malattia può associarsi alla disfunzione della tiroide. Le anomalie della coagulazione sono comuni ma raramente sono significative dal punto di vista clinico.
Altre caratteristiche includono:
- Occhi: la palpebra superiore dà l’impressione di essere pesante per cui copre parte dell’occhio (ptosi palpebrale), la rima palpebrale è rivolta verso il basso, vi è una piega cutanea che copre l’angolo interno degli occhi (epicanto) e aumento della distanza che separa i due occhi (ipertelorismo).
- Orecchie: possono presentare un impianto basso, ispessimento dell’elice ed essere ruotate posteriormente.
- Naso: la radice del naso è appiattita e questo contribuisce a far sembrare aumentata la distanza tra i due occhi.
- Bocca: filtro prominente con solcatura evidente, a volte micrognatia.
- Collo: appare corto e presenta delle pliche cutanee che dalla parte superiore del collo si estendono lateralmente sulle spalle (pterigio).
- Apparato muscolo-scheletrico: la maggior parte dei pazienti (90-95%) presenta una particolare conformazione dello sterno: carenatum (sporgente) nella parte superiore ed excavatum (depresso) in quella inferiore ed in percentuale minore degli arti superiori (cubito valgo); può essere presente cifosi o scoliosi.
- Criptorchidismo: nel 60% dei maschi vi è mancata discesa di uno o entrambi i testicoli nello scroto.
- Problemi di coagulazione: circa il 65% dei pazienti ha disturbi della coagulazione con tendenza a sanguinare facilmente.
- Pubertà: può essere ritardata (in particolare nei maschi).
- Apparato uditivo: è frequente il riscontro di otiti medie.
La Sindrome di Noonan e il Rischio di Tumori
Nell'infanzia è aumentato il rischio di sviluppare i tumori e le leucemie (in particolare la leucemia mielomonocitica giovanile), con un rischio cumulativo del 4% circa di sviluppare una neoplasia prima dei vent'anni.
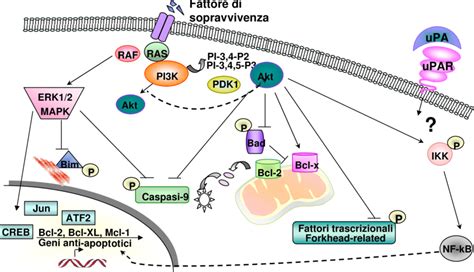
Genetica della Sindrome di Noonan: Le RASopatie
La SN è trasmessa con carattere autosomico dominante con espressività variabile e ampia eterogeneità genetica. È causata da mutazioni dei geni PTPN11 (12q24.13), presenti nel 50% dei pazienti, SOS1 (2p22.1) nel 15%, RAF1 (3p25.2), RIT1 (1q22) e LZTR1 (22q11.21) e, più raramente, di altri geni associati alla via di segnale di RAS/MAPK. La sindrome di Noonan viene ereditata nella maggior parte dei casi come carattere autosomico dominante: è sufficiente l'alterazione di una delle due copie del gene per causare la malattia. Ogni cellula del nostro corpo ha due copie di ogni gene, una copia sul cromosoma ereditato dalla madre e una copia sul cromosoma ereditato dal padre. La sindrome di Noonan si verifica quando è alterato (mutato) uno solo dei due geni che contengono le informazioni necessarie per produrre una una delle proteine della cascata RAS/MAPK. È presente una copia normale del gene che tuttavia non è sufficiente per ricostruire il messaggio che è stato danneggiato dal gene mutato.
Le RASopatie costituiscono uno tra i più comuni gruppi di patologie dello sviluppo a base non cromosomica con una prevalenza complessiva di 1/1500 nuovi nati. Fra le RASopatie, la sindrome di Noonan rappresenta la condizione più variabile dal punto di vista clinico e a più alta incidenza. Queste malattie dello sviluppo comprendono anche la sindrome cardio-facio-cutanea (CFCS), la sindrome di Costello (CS), la sindrome di Noonan con lentiggini multiple (NSML; precedentemente nota come sindrome LEOPARD), la sindrome di Mazzanti (MS), la neurofibromatosi tipo 1 (NF1), la sindrome di Legius (LS) e un numero crescente di nuove condizioni clinicamente e molecolarmente correlate. Le RASopatie condividono un meccanismo patogenetico comune caratterizzato da anomalie della regolazione della via di trasduzione del segnale mediata dalle proteine RAS.
Lo spettro clinico della SN può differire leggermente a seconda del gene-malattia interessato. È stato notato che pazienti con mutazioni diverse possono avere caratteristiche cliniche distinguibili. Per esempio, il gene PTPN11 è associato prevalentemente a stenosi polmonare valvolare, mentre il gene RAF1 causa spesso cardiomiopatia ipertrofica. Il gene SOS1 si associa nella maggior parte dei casi a statura e profilo cognitivo normali.
LISTA DEI GENI INTERESSATI E TIPO DI MUTAZIONE:
- NOONAN PTPN11: Asn308Asp, Asn308Ser, Tyr279Cys, Thr468Met, Ser502Thr, Phe285Ser, Gln79Arg, Thr411Met, Ala461Thr, Gly464Ala, Gln510Pro, Gln510Arg, Arg138Ter, Thr2Ile.
- NOONAN SOS1: Thr266Lys, Met269Arg, Arg552Gly, Arg552Ser, Trp432Arg.
- NOONAN KRAS: Thr58Ile, Val14Ile, Val152Gly, Asp153Val, Lys5Glu, Gly60Ser.
- NOONAN RAF1: Ser257Leu, Pro261Ser.
- NOONAN BRAF: Thr241Met, Thr241Arg, Trp531Cys.
- NOONAN NRAS: Gly60Glu.
- NOONAN CBL: Gln367Pro, Lys382Glu, Asp390Tyr, Arg420Gln, Tyr371His, Cys384Arg, Cys396Arg, Tyr371Cys.
In una percentuale compresa all'incirca tra un terzo e due terzi dei pazienti, la sindrome viene ereditata da un genitore affetto da sindrome di Noonan. Nei casi rimanenti, i genitori non sono affetti dalla sindrome di Noonan e si tratta quindi di mutazioni de novo il che significa che la mutazione è avvenuta durante la formazione della cellula uovo o dello spermatozoo o nelle primissime fasi di sviluppo embrionale. La mutazione de novo (50%) si verifica per la prima volta nel soggetto affetto come mutazione spontanea dell’informazione genica. È stato stabilito che la trasmissione sia autosomica dominante, sebbene sia stata recentemente identificata una forma autosomica recessiva (Johnston et al., 2018).
Diagnosi della Sindrome di Noonan
La diagnosi si basa sull'osservazione del quadro clinico, ma può essere difficile a causa della sua notevole variabilità. La diagnosi clinica di sindrome di Noonan può essere confermata mediante esame molecolare specifico. Le analisi molecolari dei geni-malattia supportano la diagnosi e la consulenza genetica. La diagnosi prenatale può essere effettuata sui villi coriali e sugli amniociti. È possibile ricercare le mutazioni del gene mediante un prelievo di sangue e studio genetico molecolare.
MED2000ECO Igroma Cistico
Strumenti Diagnostici Aggiuntivi
- Ecocardiografia fetale: per la ricerca di cardiopatie funzionali e strutturali, permettendo la diagnosi precoce delle maggiori cardiopatie congenite, anch’esse cinque volte più frequenti nella popolazione di feti affetti da igroma cistico rispetto alla popolazione generale. L’ecocardiografia fetale ha inoltre l’importante compito, in caso di non settazione dell’igroma, di identificare precocemente la stenosi polmonare, la cardiomiopatia ipertrofica e i difetti settali patognomonici della Sindrome di Noonan.
- Analisi del cariotipo: ottenibile mediante villocentesi o amniocentesi o prelievo materno per la valutazione del DNA fetale, per l’elevato rischio di aneuploidia, che risulta sei volte maggiore rispetto alla popolazione generale.
Medicina Fetale e Nuove Prospettive sulla Diagnosi Prenatale
L'approccio della medicina fetale, che riconosce al feto la dignità di paziente a tutti gli effetti, ha rivoluzionato la percezione e la gestione di patologie come l'igroma cistico. La medicina fetale pone la diagnosi prenatale come punto di partenza per cercare soluzioni terapeutiche. Studi condotti dal Professor Noia e collaboratori presso il Centro Diagnosi Terapie Fetali del Gemelli hanno dimostrato che, in una percentuale significativa di casi, l'igroma cistico non è necessariamente associato a malattie genetiche o cromosomiche con prognosi infausta. In uno studio su 220 gravidanze con diagnosi di igroma cistico, è emerso che nel 55-59% dei casi i cromosomi fetali erano normali. Ulteriori indagini, come l'ecocardiografia fetale, hanno escluso patologie cardiache nel 70% dei casi.
Questi studi hanno evidenziato che la regressione intrauterina dell'igroma cistico si osserva nel 36% dei casi, con una completa scomparsa nel 77%. Alla fine dello studio, 52 bambini sono nati sani, mentre un follow-up a lungo termine (fino a 25 anni) ha confermato che il 64% dei bambini, precedentemente a rischio di interruzione volontaria di gravidanza, era in salute. Questo sottolinea l'importanza di una consulenza genetica approfondita e di una valutazione multidisciplinare per fornire informazioni accurate e speranza alle future mamme.
Uno studio retrospettivo condotto sulla casistica di pazienti afferenti all’Hospice Perinatale - Centro per le Cure Palliative Prenatali, al Day Hospital (DH) di Ostetricia del Polo Salute della Donna e del Bambino - Policlinico “Agostino Gemelli” di Roma si propone di precisare il counseling fornito alla paziente in gravidanza a cui viene riscontrata la patologia fetale igroma cistico. Questo al fine di contenere il dubbio diagnostico e di ricercare elementi prognostici utili alla consulenza ostetrica. Lo studio retrospettivo condotto riguarda una casistica di pazienti afferenti presso l’Hospice Perinatale - Centro per le Cure Palliative Prenatali, al Day Hospital (DH) di Ostetricia del Polo Salute della Donna e del Bambino - Policlinico “Agostino Gemelli” di Roma tra il maggio 1985 e il dicembre 2012. 237 pazienti con una diagnosi di sospetta malformazione vasculo-linfatica del collo fetale sono state valutate mediante ecografie ostetriche seriate effettuate ogni tre settimane di gravidanza. Gli studi ultrasonografici sono stati eseguiti mediante Technos MP ESAOTE High Definition System e VOLUSON E8 usando sonde trans-addominali a 3,5 MHz e sonde trans-vaginali.
Una volta posta la diagnosi di certezza, la coppia (ovvero la paziente e talvolta il partner) ha ricevuto un counseling diagnostico-prognostico al fine di precisare i punti chiave della gestione ostetrica del caso, l’associazione con patologie malformative eventualmente presenti, con cromosomopatie e patologie cardiologiche fetali, e infine per illustrare l’eventualità di regressione in utero della patologia del feto, specialmente se la forma era isolata. Così è stata proposta la possibilità di precisazione diagnostica mediante accertamento del cariotipo (amniocentesi) ed ecocardiografia fetale e, in buona parte dei casi, quando questo era indicato, mediante il colloquio con un chirurgo pediatrico.
Il follow-up ostetrico è stato ottenuto mediante ripetute interviste telefoniche alle pazienti e mediante l’utilizzo del Sistema Informatico del Policlinico “A. Gemelli”. Il test di Mann-Whitney è stato compiuto per valutare l’associazione tra l’esito neonatale e l’età materna, la dimensione dell’NT e dell’igroma (diametro massimo prima e ultima misurazione) e le dimensioni dell’ascite. I neonati vivi, figli delle madri sottoposte ai predetti controlli presso il DH di Ostetricia, sono stati valutati in collaborazione con i colleghi Neuropsichiatri infantili del Dipartimento per la Tutela della Salute della Donna e della Vita Nascente, del Neonato e dell’adolescente, i quali hanno visitato i pazienti e hanno somministrato loro specifici questionari scientificamente validati al fine di valutare il grado di rilevanza degli aspetti neurocognitivi e psicomotori della patologia igroma cistico sui bambini (casistica fino ai 26 anni di età) le cui madri avevano ricevuto tale diagnosi in gravidanza. A tutti i genitori dei bambini/adolescenti/adulti con riscontro ecografico di igroma in gravidanza è stata somministrata durante un’intervista il test Vineland Adaptive Behavior Scales in forma completa per valutare la percezione dei genitori rispetto alle competenze adattive dei rispettivi figli. Nei bambini di età compresa tra 4 e i 6 anni è stato somministrato il test delle matrici progressive colorate di Raven.
Gestione Terapeutica dell'Igroma Cistico e Sindrome di Noonan
Il trattamento della patologia è sintomatico. Nei bambini piccoli è indicato un supporto nutrizionale in collaborazione con gastroenterologo e nutrizionista. Il trattamento richiede un approccio multidisciplinare. Le anomalie cardiovascolari vengono trattate secondo i protocolli standard. Il trattamento del ritardo della crescita con l'ormone della crescita rimane controverso. La prognosi è variabile dato che il quadro clinico comprende i segni clinici lievi/non evidenti nell'età adulta fino alle forme più gravi associate a cardiopatie potenzialmente letali o a tumori maligni infantili.
Terapie per l'Igroma Cistico
Il trattamento dell'igroma cistico mira a ridurre le dimensioni delle cisti e ad alleviare i sintomi associati, che possono includere problemi respiratori alla nascita se l'igroma è di grandi dimensioni e comprime le vie aeree.
- Chirurgia Resettiva: L'asportazione chirurgica della massa rimane la modalità di trattamento primaria. L'obiettivo è rimuovere completamente la cisti, anche se questo può essere complicato dalla vicinanza a strutture vitali. La recidiva è un problema comune.
- Scleroterapia: Consiste nell'iniezione di sostanze irritanti (come la bleomicina, la doxiciclina o l'OK-432) direttamente nelle cisti per indurne il collasso e la successiva fibrosi. Questo approccio meno invasivo è spesso utilizzato in pazienti non ideali per la chirurgia o come complemento ad essa.
- Terapia Laser: Un'opzione emergente, particolarmente per le cisti superficiali, che utilizza energia luminosa focalizzata per ridurne le dimensioni.
- Approccio Conservativo: In scenari specifici, quando l'igroma cistico è piccolo e asintomatico, può essere adottato un approccio di osservazione.
La Speranza dalle Cellule Staminali e dalla Terapia Prenatale
La collaborazione tra istituti di ricerca sta aprendo nuove frontiere nel trattamento delle patologie fetali, inclusi gli igromi cistici. La ricerca sulle cellule staminali mesenchimali amniotiche e sulle staminali ematopoietiche del cordone ombelicale sta esplorando il loro potenziale uso come terapia fetale per malattie genetiche. L'obiettivo è trapiantare il feto in una fase molto precoce dello sviluppo, quando la sua reazione immunitaria è ancora bassa o inesistente, per consentire la nascita di bambini sani. Studi su modelli animali hanno dimostrato la capacità di queste cellule di migrare e attecchire negli organi ematopoietici e in altri tessuti, aprendo la strada a future applicazioni terapeutiche.
Inoltre, si stanno sperimentando approcci terapeutici prenatali specifici per l'igroma cistico. Esperienze condotte inoculando sostanze come l'OK-432 direttamente all'interno dell'igroma cistico hanno mostrato una percentuale di sopravvivenza promettente. Più recentemente, nella fase postnatale, farmaci come il Sirolimus hanno dimostrato una significativa riduzione del volume di voluminosi igromi cistici.
Storia e Riconoscimento della Sindrome di Noonan
Nel 1963 Jacqueline Noonan, cardiologa pediatra all’Università del Kentucky, notò che molti bambini con un particolare difetto congenito del cuore, la stenosi della valvola polmonare, avevano alcune caratteristiche fisiche in comune, come la bassa statura, alcune alterazioni nei lineamenti del viso e una plica cutanea al collo, definita pterigio. Queste caratteristiche erano presenti sia nei maschi che nelle femmine. L’esame cromosomico delle persone affette era normale. Questa condizione fu inizialmente confusa con la sindrome di Turner, con la quale condivide la cardiopatia congenita, lo pterigio del collo e la bassa statura, ma che è dovuta ad una anomalia cromosomica (di solito assenza di un cromosoma X), in presenza di un sesso femminile. Inizialmente la sindrome descritta da Jacqueline Noonan fu definita erroneamente “sindrome di Turner maschile”. Tuttavia, nel 1965, Summit et al. riconobbero che era una condizione distinta.
Prognosi e Gestione a Lungo Termine
La prognosi dell'igroma cistico dipende in larga misura dalle sue dimensioni, dalla sua posizione e dalle eventuali complicazioni associate, in particolare la presenza di anomalie cromosomiche o malformazioni significative. Per i bambini nati con igroma cistico isolato, la prognosi a lungo termine è generalmente buona, sebbene possano essere necessari molteplici interventi chirurgici o sessioni di scleroterapia. Utilizzando trattamenti appropriati, la maggior parte delle persone con sindrome di Noonan conduce una vita adulta normale. Non esiste una prevenzione primaria per l'igroma cistico, poiché si tratta di un difetto dello sviluppo embrionale o di una condizione genetica non prevenibile tramite stili di vita o farmaci.
tags: #igroma #cistico #noonan #sigu #raccomandaziobe