L'anemia mediterranea, nota anche come beta-talassemia, è una malattia genetica ereditaria che comporta gravi conseguenze per chi ne è affetto, ma i progressi della medicina offrono oggi nuove prospettive sia in termini di trattamento che di prevenzione. In Italia, la diagnosi genetica preimpianto (PGD) rappresenta un'opzione fondamentale per le coppie a rischio di trasmettere la talassemia, consentendo loro di intraprendere una gravidanza con maggiori certezze.
Comprendere la Talassemia: Cause, Sintomi e Diffusione
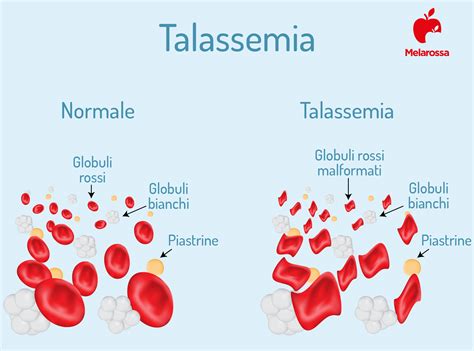
La talassemia è una malattia dovuta all’alterazione, o mutazione, di un gene chiamato HBB, che determina un’alterazione dell’emoglobina contenuta nei globuli rossi del sangue. I globuli rossi alterati sono prodotti in numero insufficiente e anche distrutti in eccesso, sicché non riescono a trasportare l’ossigeno nel sangue. Questo deficit porta a un quadro di anemia grave.
La malattia si evidenzia, in genere, a partire da qualche mese dopo la nascita con sintomi come ritardo della crescita, ittero, febbre, ingrossamento di fegato e milza, e alterazioni dello scheletro. Se non curata, può portare a morte precoce. L'unico modo per arrivare alla guarigione completa della malattia è, oggi, il trapianto di midollo osseo o di cellule staminali, preferibilmente da un familiare compatibile, ma purtroppo ciò è possibile solo nel 30% dei casi circa. La cura resta pertanto, nella maggior parte dei casi, quella classica, da continuarsi per tutta la vita: trasfusioni ogni 15-20 giorni e agenti chelanti per eliminare gli accumuli di ferro nei tessuti.
In Italia, ci sono oltre 3 milioni di portatori sani di talassemia e migliaia di talassemici. Un italiano su 20 circa è portatore sano di talassemia, cioè porta dentro di sé 1 gene sano e 1 gene “sbagliato” (mutato). Ci sono notevoli differenze fra le Regioni: i portatori sono molto più frequenti in Sardegna, nella provincia di Ferrara, in Sicilia e in genere nel Sud Italia. Se in una coppia entrambi i partner sono portatori sani, la probabilità che un figlio sia malato è del 25% a ogni gravidanza (una possibilità su quattro), mentre c'è una probabilità su quattro che il figlio sia perfettamente sano e una su due che sia a sua volta portatore.
La beta-talassemia è la forma di talassemia più grave e più diffusa in Italia, e rientra nel più ampio gruppo delle talassemie, caratterizzate da difetti nella produzione di emoglobina. L'emoglobina, essendo costituita da frammenti proteici diversi (di tipo alfa e beta), può dar luogo a due tipi principali di talassemia: alfa e beta, a seconda dei frammenti difettosi.
Esistono due forme di malattia, a seconda del tipo di mutazioni coinvolte: la talassemia major, che provoca un'anemia molto importante, e la talassemia intermedia, in cui le trasfusioni possono essere meno frequenti a seconda della gravità dell'anemia. La talassemia minor si verifica quando solo uno degli alleli porta la mutazione della malattia (un gene è sano, l’altro è beta+ o beta0).
Talassemia, cos'è e come si cura
La Diagnosi di Talassemia: Dalla Ricerca dei Portatori alle Tecniche Avanzate
La ricerca dei portatori sani di talassemia è fondamentale per prevenire la trasmissione della malattia. Per sospettare lo stato di portatore sano di talassemia è sufficiente un esame estremamente comune: l’emocromo. L'unica indicazione specifica per i portatori sani è quella di assumere periodicamente acido folico.
La diagnosi di anemia mediterranea si basa oggi su un protocollo clinico consolidato che combina l’analisi della storia familiare con esami di laboratorio di precisione. Il primo sospetto nasce solitamente da un normale emocromo. Nei soggetti con talassemia, si osserva tipicamente una microcitosi, ovvero globuli rossi di dimensioni ridotte (basso valore di MCV), associata a una riduzione dei livelli di emoglobina. Il “gold standard” per la diagnosi è l’elettroforesi dell’emoglobina o, più comunemente oggi, la cromatografia liquida ad alta prestazione (HPLC). Questi test permettono di quantificare i diversi tipi di emoglobina presenti (come l’emoglobina A2 e l’emoglobina F). L’analisi del DNA (test molecolari) rappresenta lo step definitivo e viene utilizzata per identificare le specifiche mutazioni genetiche responsabili della malattia.
Vivere con la Talassemia: Trattamenti e Qualità della Vita
Oggi, l’anemia mediterranea non spaventa più come un tempo, perché può essere tenuta sotto controllo. Il trattamento di questa malattia ha fatto notevoli progressi negli ultimi venti anni. Alle trasfusioni di sangue si accompagnano le terapie chelanti, ossia quelle terapie che riducono il ferro in eccesso causato dalle continue trasfusioni, evitando in questo modo le complicanze d’organo che l’accumulo di ferro produrrebbe. Le trasfusioni di sangue sono diventate notevolmente più sicure in termini di rischi infettivologici e il sangue viene conservato in anticoagulanti che ne mantengono una vitalità dei globuli rossi molto prolungata.
Pertanto, sia l’aspettativa di vita, con pazienti che raggiungono oltre i 60 anni di età, sia la qualità di vita, con pazienti che sono diventati nonni, sono nettamente migliorate. Una persona malata riesce a svolgere una vita tutto sommato normale, potendo andare a scuola e all'università, a lavorare, in vacanza, e anche avere figli.
L’approccio terapeutico all’anemia mediterranea ha vissuto una trasformazione radicale negli ultimi anni. Se un tempo l’obiettivo era esclusivamente la sopravvivenza, oggi la medicina punta a garantire una qualità della vita paragonabile a quella della popolazione generale e, in casi selezionati, alla guarigione definitiva.
Per i pazienti con talassemia major o forme intermedie gravi, il pilastro del trattamento rimane la terapia trasfusionale regolare. Le trasfusioni vengono solitamente effettuate ogni 2-4 settimane per mantenere i livelli di emoglobina pre-trasfusionale tra 9,5 e 10,5 g/dL. La conseguenza inevitabile delle trasfusioni croniche è il sovraccarico di ferro, per contrastarlo si utilizza la terapia chelante. I farmaci chelanti legano il ferro in eccesso e ne favoriscono l’eliminazione attraverso le urine o le feci.
Una delle innovazioni più recenti è l’introduzione di farmaci che stimolano la maturazione dei globuli rossi (agenti di maturazione eritroide come il luspatercept). Il trapianto di midollo osseo (o di cellule staminali emopoietiche) è l’opzione curativa standard, più efficace se eseguita in età pediatrica e con un donatore compatibile (solitamente un fratello).
La terapia genica rappresenta la frontiera più avanzata. Consiste nel prelevare le cellule staminali del paziente stesso, correggerne il difetto genetico in laboratorio (tramite vettori virali o tecniche di editing genomico come CRISPR/Cas9) e reinfonderle nel paziente. Lo scorso giugno, al congresso annuale della European Hematology Association, sono stati presentati i dati di uno studio di follow-up a lungo termine con la terapia genica beti-cel in pazienti con beta talassemia trasfusione-dipendente, che mirano concretamente alla guarigione.
Fertilità e Talassemia: Sfide e Possibilità
Per fertilità s’intende la capacità di avere e portare a termine una gravidanza: una ridotta fertilità è comune agli individui affetti da anemia mediterranea dipendente da trasfusioni, come la beta-talassemia maggiore. La fertilità è influenzata dalla capacità degli ovuli femminili e dello sperma maschile di maturare ed unirsi nella fecondazione. Nelle donne, è influenzata anche dalla maturazione sessuale e dalla capacità dell’utero di portare a termine una gravidanza. Un ritardo nella maturazione sessuale può escludere la possibilità di avere figli biologici finché la pubertà non sia raggiunta e, per le ragazze, fino alla comparsa del primo ciclo mestruale (menarca). Alcune donne con la beta talassemia soffrono di amenorrea primaria (cioè il ciclo mestruale non è mai iniziato), condizione che può richiedere la somministrazione di terapie ormonali. Negli uomini, lo sperma viene prodotto nei testicoli. La fertilità può essere ridotta a causa di un eccesso di ferro nella ghiandola pituitaria (ipofisi): il danno che ne deriva impedisce il rilascio di ormoni ipofisari in risposta ai segnali provenienti dall’ipotalamo.
Sembra che l’approccio migliore per cercare una gravidanza quando si ha l’anemia mediterranea consista nel tenere sotto controllo i livelli di ferro: il corpo non è in grado di sbarazzarsi delle quantità in eccesso che si accumulano per via delle trasfusioni croniche di sangue. La desferrioxamina (Desferal) aiuta a rimuovere l’eccesso di ferro. Questo farmaco viene in genere somministrato cinque notti su sette a settimana tramite una pompa che infonde lentamente la desferrioxamina sotto la pelle per diverse ore. Studi scientifici suggeriscono che un uso efficace della desferrioxamina può condurre ad una regolare maturazione sessuale. I pazienti che agiscono al meglio sono quelli che cominciano precocemente il trattamento, prima che i livelli di ferro, misurati in termini di incremento del livello di ferritina, diventino troppo elevati. Tuttavia, anche coloro con livelli di ferritina molto elevati per un lungo periodo di tempo, potrebbero avere una maturazione sessuale regolare, anche se in verità ciò accade raramente.
Anche se nelle donne affette da talassemia dipendente da trasfusione la fertilità è ridotta, in certi casi non è da escludere la possibilità di una gravidanza naturale. Donne affette da talassemia possono portare a termine una gravidanza senza rischi. Tuttavia, prima di decidere è necessario consultare il parere del proprio medico. La cura a base di DFO (desferroxamina) deve essere interrotta non appena diagnosticata la gravidanza, poiché gli effetti del medicinale sull’embrione non sono ancora del tutto chiari. Tuttavia, studi condotti sugli animali hanno dimostrato che esiste una relazione tra l’uso del farmaco e l’insorgere di gravi danni nell’embrione. Le funzioni cardiache devono essere monitorate regolarmente. In conclusione, la riproduzione in pazienti con talassemia major o intermedia è ora una realtà.
La Diagnosi Genetica Preimpianto (PGD): Un Faro di Speranza
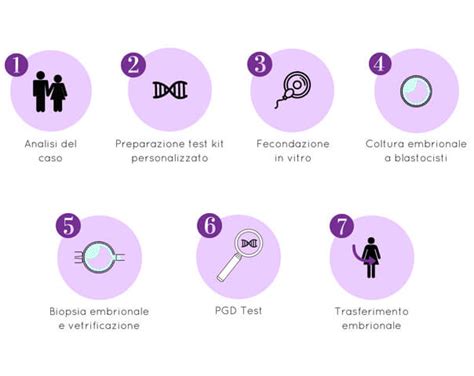
La diagnosi genetica preimpianto (PGD) è una procedura che consente alle coppie “a rischio” di malattie geneticamente trasmissibili, come la talassemia e la fibrosi cistica, di evitare il rischio che il loro bambino nasca con la malattia genetica di cui sono portatori. L’obiettivo della diagnosi genetica preimpianto è, in generale, quello di iniziare una gravidanza con degli embrioni che siano stati testati dal punto di vista genetico, nel caso specifico il gene della talassemia, al fine di identificare la presenza della malattia genetica negli embrioni generati in vitro. Così facendo, si evita l’inizio di una gravidanza con un embrione affetto, ossia un embrione che svilupperà la malattia. Pertanto, si impianterà sia l’embrione sano, sia l’embrione portatore sano della malattia.
Il percorso è il medesimo che segue una coppia infertile che accede ad un trattamento di procreazione medicalmente assistita (PMA) per permettere il recupero di ovociti da fertilizzare con gli spermatozoi del partner. Mentre nella coppia che ha un problema di infertilità l’embrione fecondato in vitro è trasferito direttamente nell’utero della donna, nel caso della coppia “a rischio genetico” l’embrione deve essere prima sottoposto a un’analisi molecolare per individuare la presenza delle mutazioni della malattia. Dopo la fertilizzazione, infatti, gli embrioni sono 'coltivati' in vitro per raggiungere, tra il quinto e il sesto giorno, lo stato di sviluppo chiamato blastocisti. A questo stadio vengono prelevate alcune cellule del trofoblasto, le prime cellule che sviluppano la placenta. Quindi non si fa un intervento diretto sull’embrione, ma sulle cellule che poi costituiranno la placenta.
Percentuali di Successo e Affidabilità della PGD
Le percentuali di successo della PGD dipendono da due fasi distinte: la fase preparativa alla diagnosi genetica di preimpianto vera e propria e la fase successiva della fecondazione in vitro. Per la prima fase, poiché per la PGD l’obiettivo è individuare gli embrioni non malati, a differenza della diagnosi prenatale che ha l’obiettivo di rilevare la presenza del feto malato, è necessario conoscere esattamente gli alleli dei pazienti, ossia la sequenza genetica del gene. Per farlo si procede con un set molecolare, chiedendo alla coppia, ma anche ai loro familiari più vicini, di eseguire un prelievo di sangue per conoscere l’allele mutato non soltanto a livello del punto della mutazione, ma anche comprendere se attorno al punto della mutazione vi sono dei polimorfismi. Riuscendo a fare un set-up molecolare appropriato, l’attendibilità della diagnosi dal punto di vista molecolare è altissima, oltre il 95%.
Per quanto riguarda la seconda fase, quella della percentuale di fecondazione, molto dipende dalla coppia. Se si tratta di una coppia giovane, tra i 20 e i 35 anni, la riserva ovarica della donna è molto buona e la probabilità di avere delle blastocisti è attorno al 50-70%. Quando l’età della donna supera i 35 anni, la resa è decisamente più bassa. Gli embrioni devono avere la possibilità di raggiungere lo stato di blastocisti e in età avanzata la probabilità che lo raggiungano si abbassa.
Possibili Complicanze della PMA e PGD
Più che di effetti collaterali, si parla di possibili complicanze. La prima riguarda la possibilità di avere una gravidanza plurima, caso in cui la patologia ostetrica aumenta notevolmente, tanto che spesso questa tipologia di gravidanze non arriva a conclusione. Ma questo aspetto con la diagnosi preimpianto viene eliminato poiché gli embrioni vengono crioconservati e trasferiti successivamente in utero uno per uno. L’altra complicanza potrebbe essere di carattere chirurgico: durante il prelievo ovocitario ci potrebbe essere uno stillicidio ematico che richiede un successivo intervento per chiudere il vaso rimasto aperto. Si tratta di casi estremamente rari, parliamo dello 0,5-0,6% dei casi. Inoltre, potrebbe esserci uno stato di discomfort della donna per l’iperstimolazione, ossia un’eccessiva produzione di follicoli dovuti proprio alla procedura della DGP.
Talassemia, cos'è e come si cura
Il Quadro Normativo Italiano e L'Accesso alla PGD
Il quadro normativo in tema di PGD è mutato negli ultimi anni. Originariamente, con la cosiddetta legge 40 - la legge del 19 febbraio 2004, n. 40 - l’accesso alle tecniche di procreazione medicalmente assistita era riservato esclusivamente alle coppie infertili. Successivamente, la Corte Costituzionale, con sentenza depositata il 5 giugno del 2015, ha dichiarato illegittimi quegli articoli della legge 40 che escludevano l’accesso alla procedura di procreazione medicalmente assistita alle coppie fertili portatrici di malattie genetiche trasmissibili certificate, ossia le cosiddette coppie “a rischio”. La Corte Costituzionale nel 2015 ha di fatto consentito l’accesso alla PGD anche alle coppie fertili a “rischio” di trasmettere una malattia genetica al proprio figlio. Inoltre, dal 2017, i percorsi di riproduzione medicalmente assistita sono entrati a far parte dei livelli essenziali di assistenza (i cosiddetti LEA), quindi teoricamente il costo della tecnica della fecondazione in vitro non dovrebbe essere a carico della coppia, ma garantita dal sistema sanitario.
Attualmente, i Centri italiani a cui le coppie possono rivolgersi sono pochi, non più di una decina di centri pubblici e alcuni centri privati. Per svolgere l’analisi molecolare, quasi tutti questi Centri si avvalgono della collaborazione di laboratori esterni. Questo comporta un “viaggio” delle cellule che vengono prima prelevate nel laboratorio di fecondazione in vitro, poi congelate e spedite nel laboratorio esterno di genetica che farà l’analisi molecolare, per poi essere rispedite indietro insieme alla comunicazione di quali siano gli embrioni malati, non malati o portatori. Questo aspetto, che avviene per mancanza di attrezzature adeguate e personale formato, può incidere sull’efficacia dell’intera procedura.
Purtroppo i costi che ricadono sulla coppia sono certamente rilevanti e non esiste un prezziario unico, nel senso che i costi nei centri privati sono molto variabili. Di recente il sottosegretario alla Salute, Pierpaolo Sileri, ha dichiarato che entro il 2021 saranno stanziati i fondi necessari per garantire la gratuità della procedura. In questo momento in Sicilia il costo della riproduzione medicalmente assistita è interamente a carico della coppia, anche nei centri pubblici.
L'Associazione Luca Coscioni è da anni in prima linea per le battaglie sui diritti civili, avendo rivolto un appello al Ministro Speranza, chiedendo di provvedere subito all’aggiornamento di questa parte dei LEA, affinché la tecnica sia erogabile a carico del Servizio Sanitario Nazionale ed accessibile nelle strutture pubbliche su tutto il territorio italiano. L’appello chiede la definizione delle tariffe per le tecniche di queste prestazioni, la previsione nei LEA delle indagini genetiche preimpianto (affinché siano considerate a tutti gli effetti parte integrante delle diagnosi prenatali), denunciando chiaramente la mancanza di strutture pubbliche in grado di erogare le prestazioni previste dalla Legge.
Secondo un elenco recentemente verificato da AICE, Associazione Italiana Centri Emofilia, i centri pubblici in grado di fornire la specifica prestazione di PGT-M per le malattie rare genetiche sono: Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico a Milano, il Servizio di Fisiopatologia della Riproduzione Umana AUSL della Romagna a Ravenna, e l'Ospedale Pediatrico Microcitemico “Antonio Cao” a Cagliari.
Un nuovo servizio volto a prevenire la trasmissione di alterazioni genetiche è disponibile da circa 6 mesi presso l’Ospedale Santa Margherita Di Cortona, Loc. La Fratta, a Cortona (Arezzo). La sede di Cortona è molto importante, in quanto punto di riferimento a livello locale e nazionale e centro pubblico, quindi accessibile a tutti coloro che ne abbiano realmente bisogno. Mediante questa tecnica, è possibile fare una diagnosi genetica sull’embrione per sapere se sia malato o sano. La tecnica PGD è già diffusa, ma fino ad oggi i cittadini italiani la potevano effettuare solo a pagamento, tranne che in due ospedali, Milano Niguarda e Cagliari Microcitemico, dove però si poteva fare per due malattie specifiche: la talassemia e la sindrome dell'X fragile. In collaborazione con l’università di Siena, si può applicare questa tecnica in tutte le coppie in cui uno dei due genitori sia portatore sano di un’alterazione genetica.

Diagnosi Prenatale e Alternative: Villocentesi, Amniocentesi e Celocentesi
La scelta di cosa fare di fronte alla possibilità di un feto malato non è semplice, è molto individuale, e dipende da vari fattori che hanno a che fare con fedi religiose, sistemi di valori, e altro ancora. Proprio perché si tratta di una scelta complessa, sarebbe bene porsi il problema ancora prima che la gravidanza inizi, per non venire colti di sorpresa e non dover prendere di corsa decisioni impegnative.
La diagnosi prenatale ha l'obiettivo di rilevare la presenza del feto malato. Se si decide di effettuare diagnosi prenatale, la tecnica d'elezione è la villocentesi. Può essere fatta prima dell'amniocentesi e permette di raccogliere il materiale più adatto per l'analisi genetica. Poiché si tratta di una tecnica invasiva, che comporta un rischio seppur minimo di aborto spontaneo, viene consigliata solo alle coppie che hanno intenzione di interrompere la gravidanza in caso di malattia del feto.
Si chiama celocentesi, ed è una nuova tecnica di diagnosi prenatale per malattie genetiche come la talassemia, messa a punto dall'équipe del dott. Aurelio Maggio, responsabile del dipartimento di ematologia dell'azienda ospedaliera Villa Sofia - Cervello di Palermo, che è anche, almeno per ora, l'unico centro italiano in cui è possibile effettuarla. La tecnica, sviluppata con il supporto fondamentale dell'Associazione Piera Cutino, prevede il prelievo di una piccolissima quantità del liquido presente in una particolare cavità, detta appunto celoma, che si forma all'interno della camera gestazionale nelle prime settimane di gravidanza, per poi lasciare il posto alla cavità amniotica. Il prelievo viene fatto per via transvaginale, sempre con controllo ecografico. Nel liquido celomatico sono presenti cellule fetali che possono essere analizzate per scoprire se il feto è affetto o meno da una particolare malattia genetica. Il grande vantaggio rispetto alla villocentesi consiste nel fatto che il prelievo può essere fatto prima, tra le sette e le nove settimane, quasi un mese prima rispetto alla villocentesi, in pratica un paio di settimane dopo aver scoperto di essere incinte. Questo offre un vantaggio temporale notevole se si pensa di interrompere la gravidanza in caso di malattia del feto. Negli ultimi 7-8 anni, il gruppo del dott. Maggio ha eseguito diverse centinaia di celocentesi, e la tecnica si è rivelata molto affidabile nell'identificazione di emoglobinopatie, come raccontato in dettaglio in un articolo scientifico pubblicato sulla rivista Praenatal Diagnosis. Tuttavia, è da sottolineare che si tratta di una tecnica invasiva, per cui ci può essere rischio di perdita fetale. Secondo uno studio pubblicato nel 2002 dal greco George Makrydimas, uno dei papà della celocentesi, il rischio di aborto associato sarebbe del due per cento, ma i dati più recenti parlano invece di un rischio più basso, intorno all'uno per cento. Questa nuova tecnica si è finora concentrata soprattutto su singoli difetti genetici, come sono appunto quelli alla base di talassemia o altre emoglobinopatie. Secondo alcuni recenti risultati, però, potrebbe anche essere in grado di offrire indicazioni su anomalie cromosomiche come la sindrome di Down, offrendo una visione d'insieme dei cromosomi del feto.
Per percorrere la strada della PGD, bisogna passare attraverso una procedura di riproduzione assistita: in questo modo si possono ottenere, in provetta, embrioni da sottoporre ad analisi, per trasferire in utero solo quelli sani o, al massimo, portatori. Così si evita il rischio di aborto terapeutico, ma va precisato che è una procedura lunga, possono volerci diversi mesi prima di arrivare all'impianto, che richiede una serie di esami e una stimolazione ormonale per la donna, e che non dà garanzie di riuscita, nel senso che l'impianto può non avvenire o la gravidanza può interrompersi precocemente. Inoltre, va sottolineato che la diagnosi dell'embrione non è una tecnica perfetta: si stima che possano esserci errori nel 3% dei casi.
Il Confronto tra PGD e Diagnosi Prenatale
In passato, l’impossibilità di fare una diagnosi sull’embrione in un Paese nel quale è possibile fare la diagnosi sul feto ai fini dell’interruzione di gravidanza era un controsenso. PGD permette di evitare l’aborto terapeutico, offrendo alle coppie una scelta più etica e meno traumatica. La sentenza della Corte Costituzionale 96 del 2015 ha permesso poi alle coppie portatrici sane di patologie genetiche trasmissibili di accedere alla procreazione medicalmente assistita dichiarando incostituzionale il divieto della Legge 40 che riservava l’accesso soltanto agli infertili. Inoltre, questa sentenza ha precisato come la possibilità di accedere alla diagnosi prima del trasferimento dell’embrione è prevista dalla stessa legge (art. 14 c. 5).
Oggi, considerando i miglioramenti della sopravvivenza dei pazienti con talassemia, si parla di malattia a prognosi aperta. Quindi la coppia “a rischio” di talassemia deve poter decidere consapevolmente se effettuare o meno la diagnosi, e per farlo è necessario che sappia che la sopravvivenza dei pazienti è più lunga, che è possibile controllare le complicanze e che è possibile sperare in terapie innovative che mirano concretamente alla guarigione.
Talassemia, cos'è e come si cura
Il ruolo della Consulenza Genetica
La consulenza genetica è sempre raccomandata in accompagnamento al test genetico per il portatore di fibrosi cistica ed è indispensabile in casi in cui uno dei partner è risultato portatore di una mutazione del gene CFTR, mentre l'altro è risultato "negativo" al test di primo livello. Il rischio che la coppia abbia un figlio con FC si stima in questa situazione intorno a 1 su 400 circa. Questo rischio naturalmente è maggiore di quello delle coppie in cui entrambi risultano negativi al test, ma è minore di quello in cui entrambi risultano portatori (che è 1 su 4 ad ogni gravidanza). Se il partner chiede un approfondimento dell'indagine genetica (test di 2° livello) e risulta negativo anche a questa, il rischio di avere un figlio con FC scende ad 1 su 1000 circa. Questo perché con un test genetico di 2° livello è possibile arrivare ad identificare circa il 90% delle mutazioni, dipende dal tipo di test che il laboratorio adotta. Il laboratorio è tenuto anche ad indicare la stima del "rischio residuo di essere portatore" quando anche a questo test il soggetto risulta negativo.
La diagnosi genetica sul feto o sull'embrione che ha un rischio di malattia FC stimato intorno a 1 su 1000 nelle strutture pubbliche non viene eseguita, perché la si ritiene indicata solo se feto o embrione hanno un rischio elevato di malattia FC (da 1 su 4 a 1 su 100 circa). Oltre che non essere indicata in base all'entità del rischio genetico, la diagnosi genetica avrebbe una parziale utilità in quanto potrebbe diagnosticare nell'embrione solo la presenza o assenza dell'unica mutazione che nella coppia è stata identificata (in questa coppia quella di cui è portatrice la madre). Quindi, la diagnosi genetica potrebbe escludere il rischio di malattia FC solo nel caso in cui diagnosticasse che l'embrione non è portatore della mutazione materna. Infatti in questa condizione, anche se il padre fosse portatore di una mutazione non riconosciuta dal test genetico e l'avesse trasmessa all'embrione, comunque non ci sarebbe la malattia FC, perché l'embrione è affetto se ha due mutazioni CFTR.
Il problema della scelta di una gravidanza è molto complesso e molto delicato e andrebbe prima di tutto "compreso" bene dalla coppia attraverso la consulenza genetica diretta con genetisti esperti di FC.