Lo sviluppo del cervello umano è un processo di straordinaria complessità, che si avvia già nelle primissime fasi della gestazione e prosegue ben oltre la nascita. Quello che accade durante il primissimo periodo di formazione della corteccia cerebrale nel feto può offrire risposte sull’insorgenza di malattie nell’età adulta, sottolineando un legame profondo tra le prime fasi dello sviluppo del cervello e la possibile comparsa di patologie neurologiche non soltanto caratteristiche dell’infanzia ma anche dell’età adulta. La gravidanza, infatti, è un periodo di grandissimi cambiamenti che interessano la donna e il bambino che porta in grembo. Lo sviluppo da un singola cellula fecondata, all’embrione al feto, è un percorso straordinario. Il cervello del bambino fa parte del sistema nervoso centrale, che comprende anche il midollo spinale.
I Fondamentali dello Sviluppo Cerebrale: Dalla Nascita dei Neuroni alla Formazione della Corteccia
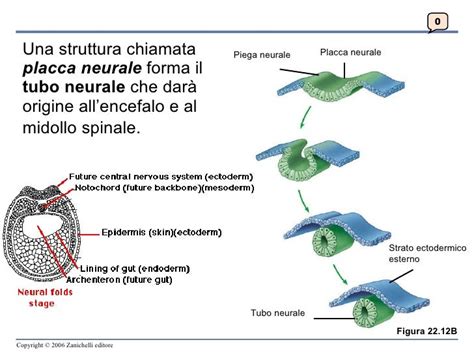
Il cervello inizia a formarsi all'inizio del primo trimestre e continua a crescere e specializzarsi. Il primo trimestre è un momento di rapido sviluppo e differenziazione delle varie parti del cervello. Entro le 4 settimane si sviluppa la struttura rudimentale nota come placca neurale, che è considerata il precursore del sistema nervoso. È in questo periodo che i neuroni e le sinapsi (connessioni) iniziano a svilupparsi nel midollo spinale. Già intorno alle 16 settimane di gravidanza compaiono due funzioni notevoli che sono la suzione e la deglutizione. La parte più attiva del cervello durante questo trimestre finale è il cervelletto, con il feto che è occupato in attività frenetiche come calci, pugni e stiramenti. Il tronco encefalico, con il suo ruolo principale e fondamentale, si occupa di mantenere in vita il corpo.
I neuroni, che costituiscono gli elementi di base (i “building blocks”) della corteccia cerebrale di un individuo adulto, sono quasi esclusivamente generati durante la vita fetale e, nella corteccia cerebrale umana, non vengono più sostituiti nel corso della vita adulta. La neurogenesi e lo sviluppo cerebrale richiedono uno stretto equilibrio tra divisione simmetrica e asimmetrica delle cellule staminali e progenitrici neurali. La corteccia cerebrale costituisce più della metà del volume del cervello umano ed è responsabile di diverse funzioni fondamentali, tra cui il ragionamento, il linguaggio e la memoria. Durante lo sviluppo embrionale, due tipi di cellule - le cellule staminali neurali e le cellule progenitrici - danno origine ai neuroni della corteccia cerebrale. A seconda della loro localizzazione, le cellule progenitrici sono classificate in progenitori apicali (AP) e basali (BP). Le AP si dividono asimmetricamente generando una AP e una BP, mentre le BP si dividono simmetricamente dando origine a due neuroni. La moltiplicazione delle cellule staminali è molto attiva durante le fasi precoci dello sviluppo cerebrale e cessa quasi completamente nella vita post-natale. L’obiettivo è capire come quello che succede nella fase di sviluppo dell’embrione e del primissimo periodo della vita post-natale possa influenzare la vita adulta.
La Connettività Neurale: Architettura Cruciale e Vulnerabilità ai Disturbi
L'accurata formazione delle reti neurali nel corso del periodo fetale è riconosciuta come essenziale per il sviluppo di un sistema nervoso sano, che non esponga il nascituro al rischio di sviluppare, dopo la nascita, numerosi disturbi neuropsichiatrici, dal disturbo da deficit di attenzione e iperattività ai disturbi dello spettro autistico, fino alla depressione maggiore e alla schizofrenia. Per tutte queste patologie sono state infatti rilevate anomalie nella connettività funzionale delle reti neurali cerebrali. La cosiddetta connettomica è uno dei campi di futura esplorazione, cercando di capire come i neuroni si definiscano e come stabiliscano connessioni tra di loro; una volta mappati i componenti cerebrali infatti è fondamentale capire come si colleghino e come scelgano i propri partner funzionali. Uno sbilanciamento in questi circuiti rappresenta il substrato cellulare di alcune malattie come epilessia, schizofrenia e autismo.
Purtroppo al momento sono ancora ignoti tempi e ordine con cui questa connettività emerge nel corso della vita intrauterina per poi stabilizzarsi. La prima mappa dello sviluppo delle connessioni cerebrali del feto in utero è stata realizzata da un gruppo di ricercatori della Wayne State University a Detroit e del National Institute of Child Health and Human Development, sempre a Detroit, che ne riferiscono in un articolo pubblicato su “Science Translational Medicine”. La tecnica, in precedenza applicata oltre che su adulti e bambini, anche su neonati e neonati pretermine, aveva già dimostrato che specifiche aree cerebrali dei due emisferi sono collegate nel periodo perinatale, inducendo a ritenere che esse iniziassero a emergere in gran parte durante il periodo di rapida crescita neurale che si verifica nel corso del terzo trimestre di gravidanza, ovvero prima dell'acquisizione di competenze cognitive. Tuttavia, queste osservazioni erano basate su studi condotti su neonati dopo gravidanze complicate.
Nella nuova ricerca, Moriah E. Thomason e colleghi hanno controllato la connettività funzionale cerebrale del feto in utero nel corso dell'ultimo periodo (dalla ventiquattresima alla trentottesima settimana di gestazione) di gravidanze non complicate, concentrandosi in particolare sulle connessioni fra i due emisferi, che attraversano il corpo calloso. Il corpo calloso è uno spesso fascio di fibre nervose che collega gli emisferi, facilitando lo sviluppo della comunicazione fra di essi e in particolare tra regioni omologhe dei due emisferi. Dall'analisi dei dati raccolti i ricercatori hanno concluso che durante il periodo esaminato era evidente una significativa connettività bilaterale in 20 delle 42 aree testate, e che la forza della connettività tra regioni cerebrali corticali omologhe aumentava con l'avanzare dell'età gestazionale. Siamo lavorando anche per capire cosa sia possibile fare per identificare queste patologie precocemente, nella speranza di individuare biomarcatori che ci guidino nella prevenzione e nell’implementazione di strategie terapeutiche precoci di numerose malattie dell’infanzia e non solo.
Una meraviglia a cui non pensiamo mai! Il Corpo Calloso, umile nastro di fibre da cui tutto dipende!
Fattori Genetici e Meccanismi Molecolari che Alterano lo Sviluppo
La genetica gioca un ruolo significativo nello sviluppo dei disturbi del neurosviluppo. Studi condotti su gemelli e famiglie hanno evidenziato che molti di questi disturbi, come l’autismo e l’ADHD, presentano una forte componente ereditaria. Ad esempio, nel caso dell’ASD, è stato dimostrato che la presenza di un fratello o una sorella con la condizione aumenta notevolmente il rischio di sviluppare il disturbo. Anche se non esiste un singolo gene responsabile, molteplici varianti genetiche possono interagire tra loro e con l’ambiente, aumentando la vulnerabilità a questi disturbi. Mutazioni in geni specifici, come quelli che regolano lo sviluppo cerebrale e la plasticità sinaptica, possono alterare i processi neurali critici durante la fase dello sviluppo. Il nostro gruppo studia come la proliferazione, la vitalità e il differenziamento dei neuroni possono essere alterati da diverse mutazioni genetiche, provocando una riduzione del volume del cervello (microcefalia) e altri disturbi del neurosviluppo.
La microcefalia può presentarsi come risultato di rare alterazioni genetiche, caratterizzate in genere da trasmissione recessiva. Citron chinasi (CITK) è una proteina che regola l’ultimo passaggio della divisione cellulare, noto come citochinesi, che porta alla separazione finale delle due cellule figlie. Il nostro gruppo ha innanzitutto scoperto in modelli sperimentali che la perdita completa di questa proteina porta ad una grave forma di microcefalia. Recentemente, il sequenziamento del genoma di pazienti affetti da microcefalia grave ha identificato mutazioni nel gene CITK, dimostrando il suo ruolo nella patogenesi della microcefalia umana. Sappiamo che i progenitori neurali degli individui portatori di mutazioni CITK non riescono a dividersi e vanno incontro a instabilità genomica e morte cellulare programmata, portando a una forte riduzione del numero di neuroni. CITK è stata originariamente identificata come una proteina importante per il rimodellamento del citoscheletro di actina.
In precedenza, è stato osservato che i cambiamenti epigenetici del DNA - modifiche reversibili del genoma che ne modificano l’organizzazione e la lettura nelle cellule, ma non la sequenza - regolano l’equilibrio tra divisione asimmetrica e simmetrica delle AP. Elena Taverna e Nereo Kalebic, Group Leaders del Centro di Ricerca in Neurogenomica di Human Technopole, in collaborazione con Tanja Vogel dell’Albert-Ludwigs-University di Friburgo (Germania), hanno affrontato questi interrogativi utilizzando tecniche avanzate di tracciamento delle linee cellulari, sequenziamento e imaging in in vitro e in vivo. I gruppi si sono concentrati sul modificatore della cromatina DOT1L che Vogel e colleghi avevano precedentemente identificato come regolatore dello sviluppo corticale nei topi. Hanno scoperto che l’inibizione farmacologica dell’attività enzimatica di DOT1L, cioè la sua capacità di modificare reversibilmente il DNA, o la riduzione dell’espressione di DOT1L aumentano la divisione simmetrica delle cellule AP e ne promuovono la differenziazione in neuroni. Per capire come l’attività di DOT1L regola la divisione e la differenziazione delle AP, i ricercatori hanno esaminato l’espressione dei geni nelle cellule AP e BP trattate con Pinometostat, un noto inibitore di DOT1L già utilizzato per il trattamento del cancro e nella riprogrammazione cellulare. L’inibizione di DOT1L modula l’espressione di diversi geni coinvolti nella regolazione del metabolismo delle cellule neuronali. In particolare, i ricercatori hanno scoperto che la riduzione dell’attività di DOT1L causa un calo dell’attività del modificatore della cromatina EZH2, che a sua volta aumenta l’espressione dell’asparagina sintetasi (ASNS), un enzima che catalizza la sintesi dell’aminoacido asparagina. DOT1L è il primo regolatore trascrizionale descritto che altera la modalità di divisione delle AP in questo modo. In sintesi, i gruppi Taverna, Kalebic e Vogel dimostrano che l’epigenetica modula l’equilibrio tra divisione simmetrica e asimmetrica nelle AP regolando il metabolismo cellulare. Nell’immagine, l’eliminazione di DOT1L nelle cellule precursori apicali (cellule verdi) modifica il modo in cui si dividono, favorendo la generazione di neuroni (cellule magenta) a scapito di altri tipi di cellule staminali nel cervello di topo in via di sviluppo.
A fronte di un costante aumento della frequenza delle patologie neurologiche di origine genetica, non sono ancora state individuate molte soluzioni per un’affidabile diagnosi preclinica e per lo sviluppo di terapie efficaci. Uno degli aspetti più limitanti in queste attività consiste nella scarsità di adeguati modelli sperimentali, capaci di stabilire un raccordo tra studi clinici e biologici. Tale problema si è reso particolarmente evidente a seguito del forte avanzamento nelle possibilità di studio genetico dei pazienti, offerto dalle moderne tecnologie di sequenziamento genomico.Modellizzare i disordini genetici in C. Il nematode Caenorhabditis elegans è un modello sperimentale estremamente potente e versatile, che permette di studiare in grande dettaglio i meccanismi molecolari delle malattie umane. Infatti, nonostante centinaia di milioni di anni di evoluzione da un antenato comune, gli esseri umani e i nematodi possiedono ancora un’alta percentuale di geni comuni, che codificano per proteine ancora più conservate. Per questo motivo, molte mutazioni umane possono essere studiate in C. elegans, permettendo di identificare molto più velocemente i meccanismi cellulari e molecolari responsabili della patogenesi della malattia. I geni dei nematodi possono essere modulati e mutati molto facilmente e le conseguenze di queste modificazioni sull’organizzazione, sulla morfologia e sulla fisiologia del sistema nervoso, nonché sul comportamento, possono essere studiate a basso costo e in un numero molto elevato di individui. Studiando come il sistema nervoso di C. elegans risponde alle mutazioni e ad altri fattori di stress possiamo rivelare i principi fondamentali della plasticità e della resilienza neurali.
L'Ambiente Materno e il Suo Impatto sul Cervello Fetale
I fattori ambientali che influiscono durante la gravidanza sono tra i più studiati nella letteratura scientifica sui disturbi del neurosviluppo. La malnutrizione è un fattore rilevante, in particolare la carenza di nutrienti essenziali, come l’acido folico durante la gravidanza o nei primi anni di vita, può influire negativamente sulla crescita e lo sviluppo cerebrale. Molte donne incinte evitano il pesce in gravidanza, per paura degli inquinanti, ma il pesce è la principale fonte di grassi omega-3. Questa preoccupazione, combinata con una dieta occidentale relativamente carente di acidi grassi omega-3, ha creato uno squilibrio di omega-3 rispetto ai grassi omega-6. Questo squilibrio può portare a uno stato proinfiammatorio che contribuisce a una serie di complicazioni, tra cui parto pretermine, ipertensione in gravidanza e depressione postpartum. Gli studi su rischi e benefici dell’integrazione degli Omega-3 in gravidanza sono discordanti.
Esposizioni a sostanze tossiche, come alcol, droghe o alcuni farmaci durante la gestazione, possono avere un impatto negativo sullo sviluppo cerebrale del feto. Anche l'esposizione a sostanze tossiche ambientali, come il piombo, il mercurio o pesticidi, può aumentare il rischio di disturbi del neurosviluppo.Altri fattori di rischio prenatali includono complicazioni durante la gravidanza: condizioni come il diabete gestazionale, infezioni materne o ipossia (carenza di ossigeno) possono compromettere lo sviluppo neurologico del bambino. Più frequentemente, la microcefalia deriva da cause ambientali come ipossia, abuso di alcool e stupefacenti o esposizione ad agenti infettivi quali rosolia, toxoplasmosi, citomegalovirus o Zika virus.
Lo stress materno da Covid-19 influisce sullo sviluppo del cervello fetale? E’ questa la domanda alla base dello studio coordinato da Catherine Limperopoulos ed espressione di tre importanti realtà cliniche di Washington DC (USA): il Children’s National Hospital, la School of Medicine and Health Sciences presso la George Washington University, e il MedStar Washington Hospital Center. Lo studio è stato condotto su 202 donne, di cui 65 soggetti sani durante la pandemia, con 92 risonanze magnetiche fetali, e 137 controlli prima della pandemia, con 182 risonanze. Le scansioni hanno permesso di determinare il volume di sei tipi di tessuto cerebrale; sono stati valutati, in particolare, il ripiegamento corticale, l’indice di girificazione locale e la profondità dei solchi. In parallelo a ogni risonanza, si è valutato il disagio materno attraverso scale convalidate di stress, ansia e depressione. Anche l’età dei genitori, con studi che hanno suggerito che l’età avanzata del padre o della madre potrebbe aumentare il rischio di autismo e altri disturbi dello sviluppo, è un fattore da considerare.

Fattori Post-Natali e Contesto Socio-Psicologico
Anche dopo la nascita, alcuni fattori possono influenzare lo sviluppo neurologico del bambino. Tra questi, la prematurità e il basso peso alla nascita sono strettamente correlati a una maggiore probabilità di disturbi del neurosviluppo, come l’ADHD o disabilità intellettive. Le infezioni nei primi anni di vita, soprattutto quelle che colpiscono il sistema nervoso centrale, possono alterare il normale sviluppo cerebrale.
Il contesto familiare e l’ambiente sociale in cui cresce un bambino possono influenzare lo sviluppo di disturbi del neurosviluppo. Fattori come la mancanza di stimolazione cognitiva o sociale nei primi anni di vita possono compromettere lo sviluppo di abilità fondamentali. L’esposizione precoce a situazioni di stress, abuso o trascuratezza (trauma o stress cronico) può alterare lo sviluppo delle aree cerebrali coinvolte nel controllo delle emozioni e nel comportamento. Le condizioni socioeconomiche, come la povertà e l’accesso limitato a cure prenatali o infantili di qualità, possono esacerbare il rischio di disturbi del neurosviluppo.
Difetti Congeniti del Sistema Nervoso: Manifestazioni e Strategie Preventive
I difetti congeniti del cervello o del midollo spinale causano una serie di problemi neurologici; alcuni possono non compromettere la salute o le capacità funzionali, mentre altri possono essere letali. Questi difetti possono presentarsi nelle prime o nelle ultime fasi dello sviluppo del feto. Tra le possibili anomalie del cervello e del midollo spinale, quelle note come difetti del tubo neurale si sviluppano entro le prime settimane di gestazione. Altri difetti, come idrocefalo e microcefalo, si sviluppano a gravidanza più avanzata. Esistono molte cause di difetti congeniti del cervello e del midollo spinale, compresi molti fattori genetici e ambientali ancora sconosciuti.Molti bambini con malformazioni del cervello e del midollo spinale presentano anomalie visibili del capo o della schiena. I sintomi di danno del cervello o del midollo spinale possono svilupparsi se l’anomalia colpisce il tessuto cerebrale o spinale. Il danno cerebrale può essere letale o dar luogo a invalidità lievi o gravi che possono includere disabilità intellettiva, convulsioni e paralisi. Il danno al midollo spinale può causare paralisi, incontinenza e perdita di sensibilità in aree del corpo raggiunte dai nervi situati sotto il livello del difetto. I sintomi tipici includono deficit intellettivo, paralisi, incontinenza o perdita di sensibilità di alcune parti del corpo.
La diagnosi si basa su varie analisi del sangue ed esami di diagnostica per immagini, come la tomografia computerizzata (TC) e la risonanza magnetica per immagini (RMI). Prima della nascita, i difetti cerebrali o del midollo spinale del feto vengono solitamente identificati con l’ecografia prenatale. In caso di risultati potenzialmente anomali all’ecografia, può essere eseguita una RMI fetale. Se viene confermata un’anomalia, i medici possono raccomandare test genetici fetali. I test genetici possono essere eseguiti mediante amniocentesi (prelievo di un campione del liquido che circonda il feto), prelievo dei villi coriali (prelievo di un piccolo campione di villi coriali, piccole proiezioni che costituiscono parte della placenta) o procedure che consentono ai medici di prelevare cellule fetali, compreso lo screening del DNA libero circolante (prelievo di un campione di sangue dalla madre da usare per identificare il DNA del feto). Dopo la nascita, TC e RMI possono rivelare difetti del cervello e del midollo spinale mediante immagini dettagliate delle strutture interne di tali organi.Se viene identificato un difetto, gli operatori sanitari forniscono ai genitori informazioni sull’anomalia e discutono le opzioni disponibili in termini di supporto psicologico e consulenza genetica, perché il rischio di avere un altro figlio con tale difetto può essere elevato.
Il trattamento dei difetti congeniti del cervello e del midollo spinale prevede spesso cure di supporto e, talvolta, un intervento chirurgico. Molti bambini con difetti congeniti del cervello o del midollo spinale necessitano di cure di supporto per assisterli nelle funzioni quotidiane, nell’istruzione e nel trattamento di complicanze o di altri difetti congeniti. Alcune anomalie, come quelle che comportano difetti di chiusura o tumefazioni visibili, possono essere riparate chirurgicamente. Anche se i danni causati dal difetto al cervello o al midollo spinale sono in genere permanenti, la chirurgia può aiutare a prevenire ulteriori complicanze e a migliorare la funzionalità. Con interventi chirurgici tempestivi, alcuni bambini hanno uno sviluppo quasi normale.
Per prevenire un difetto del tubo neurale in un feto (e nel bambino), tutte le persone che hanno in programma una gravidanza o che potrebbero essere in gravidanza devono assumere un integratore vitaminico contenente acido folico (folato), idealmente a partire da 3 mesi prima di concepire e continuandolo per tutto il primo trimestre di gravidanza. Per le donne che non hanno avuto un feto con un difetto del tubo neurale, la dose giornaliera raccomandata di folato è di 400-800 mcg (0,4-0,8 mg). Le donne che hanno avuto un figlio con un difetto del tubo neurale sono ad alto rischio di avere un altro bambino con lo stesso difetto e devono assumere una dose giornaliera più elevata di folato, 4.000 mcg (4 mg). Gli integratori contenenti folato possono non prevenire tutti i difetti del tubo neurale in gravidanze future, ma possono ridurne notevolmente il rischio.

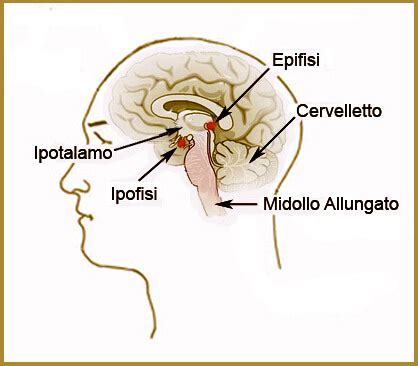
Il Craniofaringeoma: Un Esempio Dettagliato di Anomalia Sviluppata
Durante la gravidanza, nell'ipofisi dell'embrione si forma un dotto che in seguito si richiude. In alcuni bambini, tuttavia, rimangono dei residui cellulari che iniziano a crescere in modo incontrollato. È così che si sviluppa un craniofaringeoma (dal greco kranion = cranio e pharynx = faringe). Il craniofaringeoma non cresce nel tessuto cerebrale vicino e non forma metastasi (tumori figli). Tuttavia, le sue dimensioni crescenti spostano i tessuti vicini. Di conseguenza, il tumore può premere sui nervi ottici, sull'ipotalamo o sull'ipofisi, causando mal di testa, vertigini, disturbi ormonali e problemi di concentrazione. A causa dei sintomi aspecifici, il craniofaringeoma è difficile da diagnosticare. Tuttavia, quanto più precocemente il tumore viene trattato, tanto minori saranno gli effetti tardivi.
Nella maggior parte dei casi, un craniofaringeoma si manifesta in giovane età. Il 90% di tutti i craniofaringiomi si manifesta nei bambini e negli adolescenti. Un tumore cerebrale su due si forma tra il quinto e il decimo anno di vita della persona colpita. I bambini e le bambine sono rappresentati in egual misura. Negli adulti, un craniofaringeoma viene solitamente diagnosticato tra i 50 e i 75 anni. Non è ancora chiaro perché alcune persone sviluppino un craniofaringeoma. Pertanto, non è possibile prevenire questo tumore al cervello. In linea di principio, tuttavia, è consigliabile non esporre i bambini a radiazioni inutili e proteggerli dalle sostanze chimiche.
Quando le cellule del craniofaringeoma iniziano a proliferare, le persone colpite non se ne accorgono subito. Questo perché di solito crescono molto lentamente. Solo quando le strutture vicine vengono pressate dal tumore si manifestano i sintomi. Da un lato, il tumore cerebrale spesso preme sulla terza camera cerebrale (ventricolo). Ciò significa che il liquido cerebrospinale non può più circolare liberamente e la pressione intracranica aumenta. Questo provoca una sensazione di pressione alla testa e vomito, soprattutto al mattino a stomaco vuoto. Poiché i nervi ottici passano direttamente davanti all'ipofisi, il tumore spesso causa disturbi visivi, come i difetti del campo visivo. Le persone colpite non vedono più alcune cose ai margini del loro campo visivo. Nei bambini si nota che non crescono secondo la loro età o che non si verifica la pubertà con le sue caratteristiche (come i peli sul corpo). Il motivo è che gli ormoni della crescita e quelli sessuali sono prodotti dall'ipofisi. Anche l'ormone TSH, che stimola la tiroide, viene prodotto lì. Poiché il tumore altera anche l'equilibrio idrico dell'organismo, i soggetti colpiti hanno spesso una sete notevolmente elevata e di conseguenza espellono quantità di urina superiori alla media. A volte hanno anche un appetito eccessivo a causa della compromissione dell'ipofisi e mangiano molto più del necessario. Molte delle persone colpite sono quindi in forte sovrappeso. I parenti notano spesso un cambiamento psicologico nel paziente.
Se notate che voi o il vostro bambino non riuscite più a vedere tutto o avete una sete eccessiva, dovreste farvi visitare da un medico. Anche i frequenti mal di testa associati a una sensazione di pressione non devono essere ignorati, ma chiariti tempestivamente. Nel caso dei bambini, è necessario presentarsi puntualmente a tutti i controlli medici preventivi. Quanto prima il craniofaringeoma viene scoperto e trattato, tanto maggiori sono le possibilità di guarigione.
Un craniofaringeoma richiede solitamente un intervento chirurgico. Se il tumore è ancora relativamente piccolo e ben incapsulato, può essere completamente rimosso da neurochirurghi esperti senza danneggiare le strutture vicine. Tuttavia, se il tumore è già cresciuto troppo fino all'ipotalamo o ai nervi ottici, l'operazione diventa difficile. Spesso il tessuto sano e il tumore sono difficilmente distinguibili l'uno dall'altro. C'è il rischio che singole cellule tumorali rimangano e ricomincino a crescere. In questo caso è necessario un secondo intervento. Se il craniofaringeoma non è stato completamente rimosso durante l'intervento o è stato solo perforato, di solito è seguito dalla radioterapia. Questa serve a distruggere le cellule tumorali eventualmente rimaste. Il trattamento radioterapico dura di solito diverse settimane.
Se l'operazione riesce a rimuovere completamente il tumore, solo una persona su dieci tra quelle operate avrà una ricaduta nei cinque anni successivi. Tuttavia, se il tumore cresce nuovamente dopo l'intervento, il rischio aumenta nonostante una seconda operazione. In questo caso, il rischio di non sopravvivere nei cinque anni successivi è fino al 20%. A lungo termine, tuttavia, soprattutto nei bambini, la capacità di attenzione e la memoria possono essere compromesse anche se l'operazione va bene. Inoltre, in molte persone colpite i reni non funzionano più completamente. La sete eccessiva dura tutta la vita per le persone colpite. Anche il senso di sazietà e il controllo degli impulsi in generale non funzionano adeguatamente in molte persone colpite, per cui molti soffrono di disturbi alimentari e obesità, con tutte le altre conseguenze per la salute. Ai bambini devono essere somministrati ormoni della crescita dall'esterno, perché il corpo di solito non ne produce a sufficienza. Molte delle persone colpite devono anche assumere farmaci a lungo termine. In particolare, la sostituzione ormonale è spesso necessaria perché l'organismo non produce quantità sufficienti di ormoni tiroidei, della crescita o sessuali.