L'essere umano possiede un corredo cromosomico costituito da 46 cromosomi, organizzati in 23 coppie. Di questi, la metà proviene da nostro padre e l'altra metà da nostra madre, costituendo il fondamento del nostro genoma. All'interno di questi cromosomi si distribuiscono i geni, le unità ereditarie che dettano lo sviluppo e le funzioni dell'organismo. Fin dalle prime fasi embrionali, è cruciale che il numero di cromosomi sia corretto per permettere uno sviluppo con normalità. Alterazioni in questo delicato equilibrio possono portare a condizioni genetiche complesse, tra cui le trisomie, che si verificano quando è presente una copia extra di un cromosoma. Una particolare sfaccettatura di queste anomalie è il mosaicismo, una condizione in cui un individuo presenta diverse popolazioni di cellule con differenti corredi genetici.
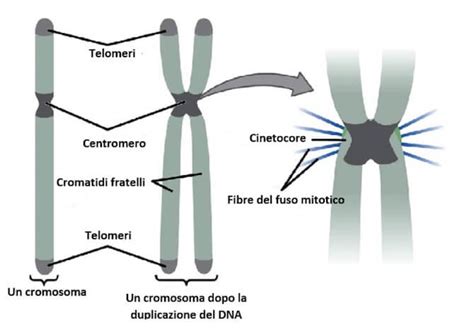
Il Corredo Cromosomico Umano e le Basi delle Alterazioni Genetiche
Ogni cellula di un essere umano sano possiede 23 paia di cromosomi omologhi: 23 di origine materna e 23 di origine paterna. Un paio di questi cromosomi è di tipo sessuale (determinando il sesso dell'individuo), mentre le restanti 22 paia sono composte da cromosomi autosomici. Nella loro totalità, i 46 cromosomi umani contengono l'intero materiale genetico, il DNA. Nel DNA di un individuo sono "scritti" i suoi tratti somatici, le sue predisposizioni e le sue doti fisiche. Ciascun paio di cromosomi contiene sequenze di DNA ben determinate, meglio conosciute come geni. Questi geni, variabili per lunghezza, producono le proteine, molecole biologiche fondamentali per la vita dell'intero organismo, attraverso processi molto precisi e coordinati.
I cromosomi possono subire diverse tipologie di alterazioni. Una riguarda parte di una loro sequenza genica (quindi un gene), e queste sono dette mutazioni, comportando solitamente la produzione di una proteina difettosa. L'altra tipologia interessa il numero di cromosomi all'interno delle cellule e sono identificate con il termine tecnico di aneuploidia. Le aneuploidie possono distinguersi per la presenza di tre copie di uno stesso cromosoma, una condizione nota come trisomia, o per quella di una sola copia cromosomica, definita monosomia. Esempi noti di trisomia includono la sindrome di Down (trisomia 21) e la sindrome di Edwards (trisomia 18), mentre la sindrome di Turner rappresenta la più importante forma di monosomia conosciuta.
La causa più comune di una trisomia è un errore genetico che può avvenire sia prima, sia poco dopo il concepimento. Di solito, al momento del concepimento, la cellula uovo (della donna) e lo spermatozoo (dell'uomo) contengono 23 cromosomi ciascuno. L'unione di questi due elementi porta alla formazione dell'uovo fecondato, o zigote, che contiene 46 cromosomi in tutto. Una trisomia insorge generalmente perché, al termine di un processo biologico molto importante chiamato meiosi, o la cellula uovo o lo spermatozoo presentano un cromosoma di troppo, quindi 24 cromosomi in tutto. Se il numero dei cromosomi è errato fin dall'inizio, l'uovo fecondato e il futuro embrione possiederanno cellule contenenti 47 cromosomi anziché 46.
Meiosi e Mitosi: I Processi Chiave
Per comprendere l'origine delle aneuploidie e del mosaicismo, è fondamentale distinguere tra meiosi e mitosi. La meiosi è il processo attraverso cui da una cellula sessuale primitiva con 46 cromosomi derivano 4 cellule sessuali mature con 23 cromosomi ciascuna. Queste 4 cellule "figlie" sono lo spermatozoo, per l'uomo, e la cellula uovo (o ovocita), per la donna. La mitosi, invece, è la divisione di una generica cellula, identificabile come "cellula madre", in due cellule figlie identiche, con un corredo cromosomico completo di 46 cromosomi. Sia la meiosi che la mitosi prevedono la duplicazione del materiale genetico (cromosomi) per garantire un'equa ripartizione del DNA in tutte le nuove cellule.
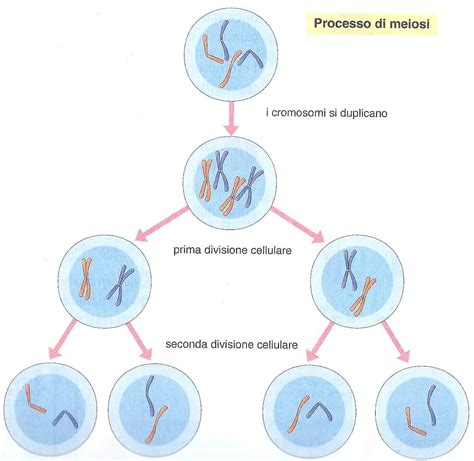
Cos'è il Mosaicismo Genetico?
Il mosaicismo genetico, o "mosaico genetico", si verifica quando in un singolo individuo sono presenti patrimoni genetici diversi, espressi contemporaneamente. Questo significa che, all'interno di una sola persona, ci sono set di cellule con un corredo cromosomico differente, oppure con lo stesso corredo espresso in modo diverso. Ad esempio, un bambino potrebbe avere un patrimonio genetico identico in tutto l'organismo, tranne che nella cute. Inutile precisare che un fenomeno del genere può avere un grosso impatto sulla salute, non sempre prevedibile. Le conseguenze dipendono da molti fattori: il numero di varianti, i tessuti interessati, i geni colpiti, e l'estensione del fenomeno. A seconda di tutti questi fattori, si possono avere casi di mosaicismo del tutto asintomatici o, al contrario, varianti che provocano deficit fisici e cognitivi.
Origini e Cause del Mosaicismo
Anche se più raramente rispetto agli errori meiotici, può capitare che l'alterazione del numero cromosomico avvenga dopo l'unione tra la cellula uovo e lo spermatozoo, cioè dopo il concepimento. In questi frangenti, i cromosomi diventano 47 quando l'embrione si sta sviluppando e sta attuando un processo di duplicazione cellulare noto come mitosi. Quando l'alterazione avviene in questa fase dello sviluppo embrionale, ha luogo il cosiddetto mosaicismo genetico. Questo termine indica che solo alcune cellule del nuovo organismo presentano 47 cromosomi, non tutte, come nel caso in cui l'errore cromosomico avveniva in meiosi.
Secondo gli studiosi, il fenomeno del mosaicismo genetico ha luogo quando l'alterazione del numero di cromosomi avviene durante la mitosi embrionale, cui è sottoposto l'uovo fecondato. In base alle ultime ricerche, sembrerebbe che il numero di cellule con 47 cromosomi dipenda da quando si verifica l'errore in fase di mitosi. Infatti, prima compare l'alterazione cromosomica e più sono le cellule dell'embrione con un numero errato di cromosomi.
Per quanto ne sappiamo, il mosaicismo si verifica durante lo sviluppo embrionale. Dopo la fecondazione, lo zigote inizia a dividersi in sempre più cellule, fino ad ottenere tutte quelle che formano un organismo umano. Qualche volta, si verifica un errore durante la divisione cellulare e si formano popolazioni di cellule con patrimoni genetici diversi. In gran parte dei casi, le cellule sbagliate muoiono e non si riproducono più, lasciando spazio a quelle con il patrimonio corretto. Altre volte, l'errore rimane ma è limitato a poche cellule. Molto più raramente, si formano interi organi con patrimoni genetici diversi, se non addirittura il 50% dell'intero organismo. Ad oggi, le cause sono poco chiare. Probabilmente, il mosaicismo è spesso frutto di anomalie sporadiche, che si manifestano casualmente e senza una vera causa. D'altra parte, può darsi che alcuni casi di mosaicismo siano causati da fattori ambientali, come l'esposizione ad agenti chimici o a radiazioni.
Mosaicismo Somatico vs. Germinale
Esistono diversi tipi di mosaicismo suddivisibili in due categorie principali:
- Mosaicismo somatico: coinvolge solo le cellule somatiche, ovvero quelle che compongono i tessuti. In questi casi, i gameti presentano un corredo genetico diverso e possono trasmettere la condizione ai figli.
- Mosaicismo germinale: Talvolta, gli individui con mosaicismo germinale non soffrono di mosaicismo somatico. Ciò significa che non mostrano alcun sintomo evidente della condizione, ma possono comunque trasmetterla. Al contrario di un individuo con mosaicismo somatico ma non germinale, che non trasmetterà la sua condizione ai figli.
Differenza tra Mosaicismo e Chimerismo
Anche il chimerismo indica la presenza di diverse linee genetiche in un solo organismo. A differenza del mosaicismo, però, il chimerismo si verifica quando due o più zigoti si fondono in un unico embrione, dando origine a un individuo con linee cellulari provenienti da zigoti diversi.
Il Mosaicismo nelle Trisomie: Le Sindromi a Mosaico
Non è detto che il mosaicismo diventi evidente: quando l’anomalia si presenta avanti nello sviluppo embrionale, può riguardare poche cellule e rimanere asintomatica per tutta la vita. Soprattutto, non è detto che l’anomalia sia dannosa per l’individuo. Tuttavia, in alcuni casi, le cellule del mosaico presentano un’anomalia cromosomica dannosa, causa di una sindrome riconosciuta. In questi casi si parla di "sindrome a mosaico". Nelle sindromi a mosaico, l’anomalia cromosomica causa della sindrome riguarda solo parte delle cellule. Ciò si potrebbe tradurre in una forma più lieve della sindrome, anche se non è detto che sia sempre così. Gli effetti del mosaicismo dipendono molto dall’estensione e dai gruppi cellulari colpiti, oltre che dal tipo di anomalia.
Trisomia 13 (Sindrome di Patau) a Mosaico
La trisomia 13, nota anche come sindrome di Patau, è una grave condizione congenita caratterizzata dalla presenza di tre copie del cromosoma 13. Nella maggior parte dei casi, i portatori di sindrome di Patau muoiono in età prenatale o entro la prima settimana di vita, rendendola una condizione generalmente mortale. Raramente, la trisomia 13 può essere limitata a un certo numero di cellule; in questo caso, si parla di mosaicismo genetico. Il 90% dei bambini che soffrono della forma completa muore pressappoco un mese dopo la nascita, mentre i bambini affetti dalla sindrome a mosaico hanno un tasso di sopravvivenza più alto, ma rimane una condizione grave.
Profilo Genetico della Trisomia 13: Negli individui con trisomia 13, il cromosoma 13 extra può essere completo (trisomia 13 totale) o parziale (trisomia 13 parziale). Nella forma a mosaico, solo alcune cellule dell'organismo presentano tre copie del cromosoma 13. In una piccola quota di casi di trisomia 13 parziale, il residuo cromosomico extra è "attaccato" a un altro cromosoma autosomico diverso dal 13, situazione nota come traslocazione. Un 10% circa dei casi di trisomia 13 parziale presenta un certo grado di mosaicismo genetico. È teoria ormai abbastanza consolidata che chi possiede un gran numero di cellule con tre copie del cromosoma 13 abbia una prognosi ancora più infausta di chi ne possiede un numero più contenuto. In altre parole, in caso di mosaicismo genetico, la gravità della malattia si accentua se l'organismo contiene un numero elevato di cellule con 47 cromosomi.
Epidemiologia e Fattori di Rischio della Sindrome di Patau: La sindrome di Patau è la terza forma di trisomia più comune al mondo, dopo la sindrome di Down e la sindrome di Edwards. I dati statistici sulla sua diffusione sono discordanti, con stime che variano da un nuovo nato ogni 8.000-12.000 a un neonato ogni 5.000. Questa incertezza è probabilmente dovuta all'alta mortalità prenatale. La trisomia 13 interessa in proporzioni uguali entrambi i sessi ed è diffusa in tutte le etnie razziali del mondo, con alcune ricerche che suggeriscono maggiori speranze di sopravvivenza per i soggetti di sesso femminile. Secondo alcune osservazioni statistiche, l'età avanzata della madre è un possibile fattore di rischio, così come la presenza di una traslocazione bilanciata, riguardante il cromosoma 13, in uno dei due genitori.
Sintomi e Segni della Sindrome di Patau: Il quadro sintomatologico dipende fortemente dalla gravità e penetranza dell'alterazione cromosomica. I portatori di una trisomia 13 totale rappresentano i casi clinici più severi. I segni e i sintomi sono numerosi e consistono in svariate alterazioni fisiche, del sistema nervoso, dell'apparato respiratorio e/o del cuore. Tra questi si annoverano:
- Testa e cervello: Testa piccola (microcefalia), oloprosencefalia (cervello non diviso in due emisferi, con problemi neurologici e difetti facciali), mielomeningocele (spina bifida aperta con protrusione di meningi e midollo spinale), fronte inclinata, naso largo.
- Orecchie e occhi: Orecchie basse e dalla forma inusuale, sordità, ricorrenti infezioni alle orecchie. Difetti oculari di vario genere come occhi molto piccoli (microftalmia) e ravvicinati (ipotelorismo), in casi gravi anoftalmia (assenza di un occhio) e/o coloboma oculare.
- Faccia e bocca: Labbro leporino (labioschisi) e/o palatoschisi.
- Pelle: Aplasia della cute (mancanza di pelle in alcune aree del cuoio capelluto), che predispone a infezioni e ulcere.
- Mani e piedi: Polidattilia (più di cinque dita), camptodattilia (flessione permanente delle articolazioni interfalangee).
- Apparato genitale: Negli individui di sesso maschile, criptorchidismo e malformazioni di scroto e genitali (micropene); nelle femmine, utero bicorne e ipertrofia del clitoride.
- Organi interni: Difficoltà respiratorie e difetti cardiaci (difetti interatriali e interventricolari, dotto arterioso pervio, valvulopatie, destrocardia), reni con cisti, anomalie a livello gastrointestinale (onfalocele ed ernia addominale).
- Sviluppo dopo il primo mese di vita: Difficoltà ad alimentarsi correttamente, costipazione, malattia di reflusso gastroesofageo, ritmo di crescita lento, scoliosi, tendenza all'irritabilità, sensibilità alla luce solare (fotofobia), ridotto tono muscolare, ipertensione, sinusiti e infezioni del tratto urinario, dell'occhio e dell'orecchio.
Sindrome di Patau e Edward - CRASH! Serie di recensioni mediche
Trisomia 18 (Sindrome di Edwards) a Mosaico
La trisomia 18, o sindrome di Edwards, è un'altra condizione molto grave. Circa il 94% dei bambini con sindrome di Edwards ha la forma completa, cioè ogni cellula del corpo possiede tre copie del cromosoma 18. Circa il 5% dei bambini con sindrome di Edwards ha la copia extra del cromosoma 18 solo in alcune cellule del corpo, nota come trisomia 18 a mosaico. La gravità dei segni e dei sintomi della malattia a mosaico dipende dal tipo e dal numero di cellule che hanno il cromosoma supplementare. Alcuni bambini possono essere colpiti solo lievemente, mentre altri lo sono in modo molto grave. La forma a mosaicismo rimane una condizione seria, ma si associa a un tasso maggiore di sopravvivenza rispetto alla forma completa.
Studi sul decorso naturale della trisomia 18 e 13, condotti tramite questionari e registri medici su 98 famiglie con trisomia 18 e 32 con trisomia 13, hanno rivelato importanti dati. La sopravvivenza in questo gruppo di bambini è stata migliore rispetto ad altri studi, sebbene fosse chiaro che queste condizioni sono gravemente disabilitanti e il tasso di morte prematura è alto. L'età media materna e paterna alla nascita mostra una distribuzione non bimodale. La distribuzione dell'età gestazionale alla nascita è diversa dalla norma della popolazione per la trisomia 18, ma non per la trisomia 13. Il peso medio alla nascita era simile tra i dati genitoriali e medici.
Complicanze della gravidanza osservate per la trisomia 18 includevano 29 casi con ritardo della crescita intrauterina (IUGR) e 17 casi con polidramnios (eccesso di liquido amniotico). Altre complicanze erano estremamente varie. Le anomalie più comuni notate con gli ultrasuoni erano IUGR (17 casi, molti nel terzo trimestre) e polidramnios (10 casi). L'incidenza di una singola arteria ombelicale (SUA) riportata era del 38% per la trisomia 18 e del 28% per la trisomia 13. I risultati placentari menzionati nei casi di trisomia 18 includevano placente piccole (18 casi), cordone piccolo, corto o sottile (10 casi), inserzione eccentrica del cordone (2 casi), ciste del cordone (3 casi), calcificazioni della placenta (2 casi) e distacco (2 casi).
Trisomia 8 a Mosaico
La trisomia 8 a mosaico è un'anomalia autosomica rara, caratterizzata dalla presenza di tre copie del cromosoma 8 in alcune cellule dell'organismo. Storicamente, la prevalenza alla nascita era stimata in 1/25.000-50.000, sebbene manchino studi epidemiologici a supporto di tale stima. La trisomia 8 in mosaico è causata da un evento post-zigotico (errore nella segregazione cromosomica durante la mitosi in un feto con cariotipo normale, o è secondario alla correzione spontanea della trisomia 8). La trisomia 8 completa, causata da un errore nella segregazione cromosomica durante la meiosi, spesso provoca un aborto spontaneo durante il primo trimestre. Quando, eccezionalmente, il feto sopravvive, presenta lo stesso fenotipo della trisomia in mosaico. La presa in carico si basa su un approccio multidisciplinare.
I dismorfismi facciali sono lievi e comprendono l'allungamento del cranio (scafocefalia), la fronte prominente, l'ipertelorismo, gli occhi infossati (50%), il naso globoso con la punta rivolta verso l'alto (60%), la micrognazia (con labbro inferiore anteverso), le orecchie grandi e displastiche con antelici prominenti e lobi grandi. I pazienti possono presentare anche agenesia del corpo calloso, palato ogivale o palatoschisi (8%), collo corto, allungamento e restringimento del torace, delle spalle e del bacino. Sono comuni le anomalie urinarie (idronefrosi, reflusso ureterale), le cardiopatie e i difetti dei grandi vasi (rispettivamente 40% e 25%). Sono comuni anche la camptodattilia (70%), l'artrogriposi delle articolazioni (che si aggrava con il tempo), le pliche palmari profonde (nei neonati) e i solchi plantari profondi (75%), l'agenesia o l'ipoplasia delle rotule, le malformazioni vertebrali (65%; anomalie della segmentazione e delle costole, scoliosi), l'epilessia, le opacità corneali e lo strabismo. La maggior parte dei pazienti presenta disabilità intellettiva lieve-moderata (QI compreso tra 50 e 75), mentre in alcuni l'intelligenza è normale. Di solito si osserva un ritardo che colpisce maggiormente il linguaggio rispetto ad altri ambiti dello sviluppo.
La trisomia 8 in mosaico sembra predisporre ai tumori di Wilms, alle mielodisplasie e alla leucemia mieloide. Alcuni pazienti affetti da trisomia 8 in mosaico hanno avuto figli.
Mosaicismo dei Cromosomi Sessuali
Un altro tipo di anomalie relativamente comuni sono quelle che interessano i cromosomi sessuali, X e Y. Proprio come nel caso delle trisomie, il mosaicismo esiste anche per le anomalie dei cromosomi sessuali.
- Sindrome di Turner a mosaico: Le donne affette da sindrome di Turner hanno una sola copia del cromosoma X, al posto della coppia di cromosomi X che dovrebbero avere. In realtà, più della metà di loro soffre della versione a mosaico della sindrome. Il mosaicismo riduce la severità di alcuni sintomi, tra cui l’infertilità: solo il 15% delle donne affette dalla sindrome riesce ad avere una gravidanza spontanea, e tutte quelle che ci riescono sono affette da mosaicismo.
- Sindrome di Klinefelter a mosaico: Chi ne soffre ha un cromosoma X extra, il che provoca sintomi come basso testosterone e fibrosi testicolare. Anche in questo caso, il mosaicismo riduce la gravità della sindrome.
Diagnosi Pre-Impianto e Mosaicismo Embrionale
Nel contesto della fertilizzazione in vitro, è possibile prevenire situazioni di aneuploidia e mosaicismo mediante una diagnosi cromosomica completa dell’embrione (PGS/PGT-A/CCS). Le potenti tecnologie attuali, come array-CGH o Next Generation Sequencing (NGS), permettono di individuare eventuali anomalie nel numero di qualsiasi cromosoma e di trasferire quindi solo gli embrioni cromosomicamente sani. Questa diagnosi viene effettuata mediante una biopsia dell’embrione, che consiste nell’estrarre da 5 a 10 cellule dallo strato esterno dell’embrione (futura placenta) tra il 5º e il 7º giorno dello sviluppo embrionale. Questa procedura non ostacola il corretto sviluppo e annidamento dell’embrione.
Il mosaicismo embrionale consiste in un embrione con una serie di cellule cromosomicamente normali e anormali (per uno o vari cromosomi). Questa anomalia è dovuta a una separazione incorretta dei cromosomi durante la divisione dell’embrione e non sembra essere associata a nessun fattore materno o paterno. Stando agli studi previ, si stima che circa un 20% degli embrioni umani presentano mosaicismo cromosomico.
È stato dimostrato che gli embrioni diagnosticati con mosaico hanno una capacità di annidamento e di gravidanza evolutiva leggermente inferiore rispetto agli embrioni privi di mosaicismo, ma questi non sono dati allarmanti. Secondo lo studio di Lledó et al. (2017), questi embrioni danno origine a un maggior numero di aborti precoci rispetto agli embrioni normali e circa un 30% sfociano in gravidanze normali. A quanto pare, questi embrioni hanno la capacità di correggere in un certo modo le cellule anomale o semplicemente queste cellule si dividono più lentamente rispetto al resto e alla fine scompaiono, dando origine a una gravidanza normale.
Nel caso del trasferimento di embrioni mosaico, si dovranno tenere in considerazione e essere trasferiti solo in assenza di embrioni normali, e ai pazienti dovrà essere sempre fornito un consiglio genetico previo al trasferimento. È importante informare del tipo di mosaicismo e del rischio specifico che potrebbe esistere in ciascun caso (problemi di annidamento, aborto o bambino con qualche sindrome o malformazione). Fino ad ora, di tutti gli embrioni mosaico trasferiti a livello mondiale non si è verificata nessuna nascita di bambini con problemi associati a mosaicismo diagnosticato nell’embrione. La Società Internazionale di Diagnosi Genetica Pre-Impianto (PGDIS) consiglia di effettuare un'amniocentesi per confermare che il cariotipo del feto sia normale o almeno un test prenatale non invasivo (NIPT).
Il Mosaicismo e la Diagnosi Prenatale
Il sospetto di mosaicismo può emergere in diversi momenti del percorso diagnostico prenatale, che include una serie di esami di screening e diagnostici. Tra gli strumenti diagnostici, l'ecografia fetale può rivelare anomalie strutturali. Il test di screening combinato, che di solito si effettua tra la 11a e la 14a settimana di gestazione, comprende un esame specifico del sangue e il test della translucenza nucale (un'ecografia mirata per la ricerca dell'ispessimento cutaneo nella parte posteriore del collo del bambino). Recentemente, è stato messo a punto un test che prevede l'analisi del DNA fetale (screening prenatale non invasivo basato sul DNA), ottenuto da un campione di sangue materno dalla 10a settimana in avanti, su cui è possibile effettuare esami citogenetici e molecolari. Le indagini principali per lo studio dei cromosomi fetali e la diagnosi prenatale delle aneuploidie rimangono la villocentesi e l'amniocentesi.
Test Prenatale Non Invasivo (NIPT)
Il NIPT è un test di screening, non diagnostico. Analizza frammenti di DNA libero circolante (cfDNA) nel sangue materno, che provengono prevalentemente dalla placenta, non direttamente dalle cellule fetali. La causa più comune di falso positivo al NIPT è il mosaicismo confinato alla placenta (CPM). La presenza di un gemello non vitale (vanishing twin), anche se riassorbito precocemente, può rilasciare cfDNA anomalo in circolo per settimane, alterando il risultato. Su questo punto le società scientifiche sono concordi e inequivocabili. È importante sottolinearlo: un NIPT positivo non significa che il feto sia affetto. Significa che è necessario approfondire. L'interpretazione del DNA fetale circolante richiede familiarità con le possibili cause biologiche di discordanza, come mosaicismo confinato alla placenta, vanishing twin o anomalie cromosomiche a bassa percentuale. L’analisi del cfDNA prevede algoritmi statistici complessi che stimano la frazione fetale, la distribuzione delle sequenze cromosomiche e il rischio di aneuploidia.
Villocentesi (Prelievo dei Villi Coriali)
La villocentesi è una tecnica per la diagnosi prenatale, più invasiva dell'amniocentesi, che si esegue più precocemente, intorno alla 10a-11a settimana. Consiste nel prelievo per via transaddominale, e sotto controllo ecografico, di tessuto trofoblastico, contenente cellule fetali. Sul campione si può eseguire un'analisi cromosomica diretta o, come nel caso del liquido amniotico, le cellule sono poste in coltura e successivamente analizzate. Quando si esegue una villocentesi, il laboratorio analizza due componenti distinte del villo coriale: il citotrofoblasto, esaminato con il metodo diretto, e la componente mesenchimale, analizzata attraverso la coltura cellulare. Questa distinzione non è solo accademica. Un mosaicismo rilevato nel citotrofoblasto ma non nel nucleo mesenchimale del villo è generalmente un CPM; se invece è presente nel mesenchima, potrebbe essere sia confinato alla placenta che generalizzato.
Il mosaicismo cromosomico nei villi coriali viene rilevato in circa l’1-2% dei casi. Quando si riscontra questa condizione è necessario un cariotipo di conferma su amniociti per distinguere un CPM da un vero mosaicismo fetale. In una grande casistica di oltre 15.000 CVS, la linea cellulare anomala si è estesa al feto nel 12,8% dei casi in cui era stato rilevato mosaicismo ai villi.
Amniocentesi
L'amniocentesi rimane l'indagine principale per lo studio dei cromosomi fetali e la diagnosi prenatale delle aneuploidie. Eseguita intorno alla 16a-18a settimana, consente il prelievo del liquido amniotico dalla cavità uterina attraverso la parete addominale. Il liquido amniotico è il fluido che avvolge e protegge il feto durante il suo sviluppo uterino. Le cellule ottenute dall’amniocentesi originano dall’epiblasto della massa cellulare interna, e sono quindi più direttamente rappresentative dell’embrione rispetto a quelle della villocentesi. L’amniocentesi analizza gli amniociti, cellule di origine fetale presenti nel liquido amniotico.
Anche nell'amniocentesi, non tutti i risultati che sembrano indicare un mosaicismo hanno lo stesso significato clinico. Si distinguono diversi livelli:
- Livello I: viene osservata una singola cellula anomala (pseudomosaicismo a cellula singola).
- Livello II: due o più cellule anomale con la stessa anomalia cromosomica compaiono in una singola coltura o in una singola colonia, ma non in colture indipendenti (pseudomosaicismo a cellule multiple).
- Livello III: cellule anomale con la stessa anomalia cromosomica sono presenti in due o più colture indipendenti (vero mosaicismo).
I dati provenienti da grandi casistiche sono chiari. Nelle amniocentesi eseguite prevalentemente per età materna avanzata, circa il 3,73% dei casi presenta pseudomosaicismo a cellula singola (livello I), lo 0,76% pseudomosaicismo a cellule multiple (livello II), e solo lo 0,2% un vero mosaicismo (livello III). Lo pseudomosaicismo è uno dei limiti tecnici riconosciuti dell’analisi citogenetica prenatale. Non riflette la reale costituzione cromosomica del feto, ma può originare da errori nella divisione cellulare durante la coltura in laboratorio, o in alcuni casi dalla presenza di cellule di origine placentare o materna nel campione. Quando si rileva un livello III, o quando un livello II suscita dubbi clinici, la diagnosi non si ferma al cariotipo classico. Il vero mosaicismo fetale riguarda più frequentemente anomalie dei cromosomi sessuali o trisomie dei cromosomi 21, 18 e 13.
Mosaicismo Confinato alla Placenta (CPM)
Il mosaicismo fetale e placentare e il mosaicismo confinato alla placenta possono influenzare il risultato del test del DNA fetale. Nel percorso della diagnosi prenatale esistono condizioni biologiche complesse che possono rendere l’interpretazione dei risultati più articolata. Il CPM si riscontra nel 2-3% delle gravidanze. I cromosomi più frequentemente coinvolti nel mosaicismo (oltre 25 casi nelle grandi casistiche) sono 2, 7, 8, 13, 18 e 21; i cromosomi 3, 9, 15, 16 e 20 mostrano un coinvolgimento intermedio; trisomie dei cromosomi 1, 4, 5, 6, 10, 11, 12, 14, 17, 19 e 22 sono le più rare. È importante sottolineare che anche quando il mosaicismo rimane confinato alla placenta e il feto è cromosomicamente normale, il CPM non è privo di conseguenze.
In alcuni casi, l’anomalia cromosomica non riguarda il feto, ma solo la placenta. Si parla di mosaicismo confinato alla placenta (CPM) quando una linea cellulare cromosomicamente anomala è presente esclusivamente nella placenta, mentre il corredo cromosomico del feto è normale. Per capire perché il CPM possa influenzare il NIPT, è utile chiarire un aspetto spesso frainteso: il DNA circolante nel sangue materno analizzato con il NIPT non proviene direttamente dalle cellule del feto, ma dall’apoptosi, cioè dalla normale morte cellulare delle cellule del trofoblasto placentare. In pratica, il test “legge” la placenta, non il feto. Scoprire che la propria placenta presenta un mosaicismo cromosomico può generare preoccupazione, ed è giusto sapere che in alcuni casi questa condizione non è del tutto priva di implicazioni. Pur essendo nella maggior parte dei casi associato a esiti fetali normali, il CPM è stato correlato in alcuni casi a restrizione della crescita intrauterina, perdita della gravidanza o morte perinatale.

Aspetti Clinici e Ricerca sul Mosaicismo
Le condizioni di discordanza tra test di screening e test diagnostico rappresentano una minoranza dei casi, ma sono cruciali da comprendere. Una delle discordanze cliniche più frequenti si presenta quando il NIPT risulta ad alto rischio ma l’ecografia è normale: in questi casi il mosaicismo, soprattutto confinato alla placenta, è una delle spiegazioni biologiche più plausibili. Analogamente, la discordanza tra villocentesi e amniocentesi può verificarsi proprio in presenza di mosaicismo feto-placentare, dato che la villocentesi esamina le cellule del citotrofoblasto e del sinciziotrofoblasto placentare, mentre l’amniocentesi analizza gli amniociti di origine fetale. Quanto più basso è il rapporto di mosaicismo rilevato al NIPT, tanto maggiore è la probabilità che il risultato sia discordante rispetto all’esito del test diagnostico invasivo. Anche valori anomali di marcatori biochimici del primo trimestre (come PAPP-A ridotta) in presenza di un NIPT rassicurante e un’ecografia apparentemente normale possono talvolta essere un segnale indiretto di mosaicismo.
In un contesto biologico complesso come quello del mosaicismo fetale o placentare, la qualità del laboratorio e dell’interpretazione dei dati assume un ruolo centrale. La consulenza genetica rappresenta un passaggio chiave. In presenza di condizioni come il mosaicismo, la comunicazione corretta è parte integrante della qualità del test.
Il Ruolo della Placenta nel Mosaicismo
Cosa succede se il mosaicismo interessa la placenta, un organo fondamentale per lo sviluppo del feto? Spesso non succede niente: nonostante il mosaicismo placentare, le cellule del feto rimangono normali, senza anomalie cromosomiche. Un team di ricerca dell’Università di Cambridge ha approfondito questa tematica. I ricercatori hanno analizzato le mutazioni presenti in 86 placente, eseguendo numerose micro-dissezioni per prelevare il DNA da diversi punti. Analizzando i tessuti, hanno scoperto un numero elevatissimo di anomalie cromosomiche, molto più elevato di quelle che si possono riscontrare nei tessuti embrionali. Il mosaicismo placentare riguarda circa il 2% dei campioni ottenuti con la villocentesi tra la 10a e la 12a settimana. Ciononostante, solo il 10% di quegli embrioni riporta quegli stessi difetti cromosomici. Benché le implicazioni non siano ancora del tutto chiare, può darsi che una delle funzioni della placenta sia “spazzare via” le anomalie cromosomiche. Si potrebbe vedere la placenta come uno spazzino di difetti genetici. Inoltre, può darsi che la placenta abbia semplicemente una soglia di controllo delle mutazioni più bassa: la placenta deve adattarsi rapidamente, il che aumenta il rischio di errori genetici. Il DNA anomalo della placenta, infatti, può essere scambiato per DNA anomalo fetale e portare a falsi positivi negli esami prenatali. Il rischio è particolarmente rilevante per le trisomie, le anomalie cromosomiche più rilevate dai test del DNA fetale. Ecco perché si eseguono sempre ulteriori controlli, dopo un esito positivo del test.
Rischi per lo Sviluppo Fetale Legati al Mosaicismo Placentare
Un mosaicismo placentare esteso, o riguardante tessuti placentari critici, può comunque influenzare negativamente lo sviluppo fetale. Pare infatti esserci una correlazione tra mosaicismo placentare significativo e:
- Restrizione della crescita intrauterina, dovuta forse a una riduzione dell’apporto di nutrienti e ossigeno al feto.
- Complicazioni della gravidanza, come preeclampsia e parto pretermine.
Mosaicismo nelle Cellule Staminali
Il mosaicismo può riguardare anche le cellule staminali. Pare che il midollo osseo di 1 persona su 40 mostri segni di mosaicismo, senza però problemi di salute evidenti. Ciò significa che il mosaicismo potrebbe essere molto più comune di quanto si creda, anche se non sempre evidente. È stato perfino documentato il caso di una donna affetta da sindrome di Turner a mosaico che ha donato il suo midollo osseo. Per quanto riguarda le staminali del cordone ombelicale, gli studi mostrano una certa stabilità genetica.
In sintesi, il mosaicismo è un fenomeno strettamente legato al mondo della genetica e delle anomalie cromosomiche, che potrebbe complicare la diagnosi di malattie più o meno rare. L'unico metodo per avere una diagnosi sicura del mosaicismo sono i test genetici e i test di screening prenatale. Si analizzano le cellule del feto e si verifica l'eventuale presenza di cellule con anomalie cromosomiche. In caso di esito positivo, si prosegue con ulteriori test genetici. Gli effetti del mosaicismo variano molto in base alla gravità e alle cellule interessate. In generale, è difficile prevedere con sicurezza gli effetti di una condizione del genere. Una bassa percentuale di cellule anomale può non avere conseguenze sull'individuo, ma avere ripercussioni sulla sua prole, mentre un'anomalia cromosomica presente nella maggior parte delle cellule è quasi equivalente a un'anomalia presente nel 100% delle cellule.
tags: #fase #del #mosaicismo #trisomia