Introduzione: Comprendere la Sindrome di Down
La Sindrome di Down, scientificamente nota come Trisomia 21, rappresenta una condizione genetica che costituisce l’anomalia cromosomica numerica più frequente tra i soggetti nati vivi. Questa condizione è dovuta alla presenza di un cromosoma soprannumerario, nello specifico il cromosoma 21, il quale è presente in tre copie anziché le usuali due in tutte le cellule dell’organismo o in una parte di esse. Proprio per questa peculiarità, la sindrome prende anche il nome di trisomia 21. Si tratta di una condizione che accompagna la specie umana, manifestandosi senza differenze tra i sessi e le etnie.
Il nucleo di ciascuna cellula umana contiene normalmente 46 cromosomi, suddivisi in 23 coppie, dove ogni coppia è formata da un cromosoma ereditato dalla madre e da uno ereditato dal padre. Nelle persone con Sindrome di Down, i cromosomi presenti non sono più 46, ma 47, in quanto il cromosoma 21 è presente tre volte. Questa anomalia cromosomica è la causa più comune di ritardo mentale nell’uomo, oltre a comportare una serie di caratteristiche fisiche distintive e una maggiore suscettibilità a determinate problematiche di salute.
L'incidenza di nati vivi con la sindrome a livello mondiale è di circa uno su 1.000, mentre l’incidenza varia, a seconda della zona geografica, da 1 su 319 bambini a 1 su 1.000. Negli Stati Uniti, l'incidenza complessiva è di circa 1/700 nati vivi. In Italia, secondo alcune stime, circa un bambino ogni 1.200 nati ha la sindrome: si tratterebbe di circa 500 nascite all’anno per un totale stimato di 38.000 persone nel Paese. Queste statistiche variano nelle diverse regioni del mondo a seconda delle politiche che riguardano la diagnosi prenatale, dei livelli di assistenza offerti alle persone disabili e alle loro famiglie, e delle leggi sull’aborto.
È essenziale utilizzare una terminologia rispettosa e aggiornata. Espressioni storiche come “mongolismo” o “mongoloide” sono considerate inappropriate e stigmatizzanti e non devono essere usate, in linea con l'obiettivo di favorire una maggiore inclusione nella società.
Capitolo 1: La Nomenclatura e la Scoperta
La Sindrome di Down deve il suo nome al medico inglese John Langdon Down, il quale, nel 1866, fu l’autore di una pubblicazione scientifica in cui descriveva in maniera completa e approfondita la sindrome, identificandone per primo i tratti comuni nelle persone affette. John Langdon Down aveva incontrato per la prima volta una ragazza con disabilità dello sviluppo quando lei aveva circa 18 anni, un incontro che lo colpì a tal punto da spingerlo a studiare medicina e dedicare la propria vita alla cura dei disabili. Nella sua descrizione, Down fece riferimento a caratteristiche fondamentali quali una faccia ampia, pieghe epicantali (cioè attorno agli occhi), lingua grossa, modesta riduzione del tono muscolare, difficoltà linguistiche e una durata della vita più breve. Segnalò inoltre la presenza di una personalità “umorale”, ovvero poco prevedibile, e il fatto distintivo che nei soggetti con Sindrome di Down i progressi ottenuti sul piano dell’apprendimento sono instabili, nel senso che possono essere facilmente perduti. Sebbene alcune di queste osservazioni fossero basate sui limiti delle conoscenze dell'epoca, la sua acuta capacità di osservazione pose le basi per gli studi futuri.

Nel corso del tempo, la comprensione di questa condizione genetica si è evoluta notevolmente, superando le iniziali descrizioni e approfondendo le basi biologiche e le complessità cliniche. L'identificazione della presenza del cromosoma 21 soprannumerario ha segnato un passo fondamentale, trasformando la sindrome da un'entità puramente clinica a una con una chiara eziologia genetica.
Capitolo 2: Le Basi Genetiche della Trisomia 21
La Sindrome di Down è causata dalla presenza (parziale o totale) di una terza copia del cromosoma 21. Questa anomalia deriva da un processo di divisione cellulare atipico al momento del concepimento. Nelle cellule umane, ci sono 46 cromosomi divisi in 23 coppie numerate; nella Sindrome di Down, il cromosoma 21 è presente tre volte anziché le normali due. Sebbene la presenza di una porzione in più del cromosoma 21 sia l'unica certezza genetica, le cause esatte che portano a questa trisomia sono ancora in parte ignote.
La Trisomia 21 "Libera" o da Non Disgiunzione: La Causa Più Comune
In circa il 95% dei casi, la Sindrome di Down è causata da una non disgiunzione, un fenomeno che porta alla formazione di un cromosoma 21 supplementare, che è tipicamente di origine materna. La non disgiunzione si riferisce al mancato corretto distacco dei cromosomi omologhi o dei cromatidi fratelli durante la meiosi dei gameti. Questo significa che, al momento della formazione dell'ovulo o dello spermatozoo, una delle cellule germinali riceve due copie del cromosoma 21 invece di una. Quando questa cellula si unisce a una cellula germinale normale, il nascituro avrà tre copie del cromosoma 21. Questa è la modalità più diffusa per la nascita di un bambino Down, e si parla in questo caso di Trisomia 21 "libera", poiché il cromosoma "in più" fluttua libero all'interno delle cellule.
La Traslocazione: Una Variante Meno Frequente ma Significativa
Circa il 4% dei casi di Sindrome di Down è dovuto a una traslocazione genetica. In questo scenario, il cromosoma 21 "in più" è legato ad un altro cromosoma, solitamente il cromosoma 14, il 22 o un altro cromosoma 21. In una traslocazione bilanciata, il materiale genetico viene scambiato con materiale proveniente da un altro cromosoma non-omologo, e la conta cromosomica viene mantenuta a 46. Tuttavia, nella Sindrome di Down, si verifica una traslocazione sbilanciata, che comporta un guadagno o una perdita di materiale genetico, creando uno squilibrio e successive anomalie cliniche.
La traslocazione più comune è la t(14;21), in cui il cromosoma 21 è attaccato al cromosoma 14; questa è una traslocazione sbilanciata, con una conta cromosomica di 45, ma con il materiale genetico del cromosoma 21 presente tre volte. In circa la metà dei soggetti con t(14;21), entrambi i genitori hanno un cariotipo normale, mostrando una traslocazione insorta de novo. Nell'altra metà, un genitore (quasi sempre la madre), che non ha la Sindrome di Down, ha solo 45 cromosomi, uno dei quali è t(14;21) in forma bilanciata, cioè è un portatore sano.
Il rischio di ricorrenza per i genitori varia. Se una madre è portatrice di una traslocazione Robertsoniana (es. t(14;21)), la probabilità di avere un bambino con Sindrome di Down è di circa 1:10 (o 10-15%). Se il padre è il portatore, il rischio è inferiore, solo di circa 1:20 (o 1-3%). Un'altra tra le traslocazioni più frequenti è la t(21;22), con rischi simili. La traslocazione 21q21q, che si verifica quando il cromosoma 21 è collegato a un altro cromosoma 21, è molto meno comune. In questo caso, un genitore portatore ha una probabilità del 100% di avere un figlio affetto dalla Sindrome di Down, poiché tutta la prole vitale avrebbe la sindrome di Down o la monosomia 21, condizione tipicamente incompatibile con la vita. Per questo motivo, è fondamentale determinare se un genitore è portatore o mosaico per la traslocazione 21q;21q per comprendere i rischi per i loro figli.
Il Mosaicismo: Una Condizione Complessa
Un altro fenomeno che può causare l’insorgere della trisomia 21, molto più raro, è quello del "mosaicismo". Esso si manifesta in circa il 2-3% (o 3%) dei casi e presumibilmente risulta da non-disgiunzione durante la divisione cellulare nell'embrione, non manifestandosi in tutte le cellule dell'individuo, ma solo in quelle che provengono dalla riproduzione della cellula mutata. Le persone con mosaicismo della Sindrome di Down hanno due linee cellulari: una con i normali 46 cromosomi e una con 47 cromosomi, tra cui un cromosoma 21 in più, o un diverso numero di cromosomi, a seconda della traslocazione.
Nelle persone con mosaicismo, il quadro clinico può essere più sfumato in funzione dell'entità del mosaicismo. Sebbene il loro rischio di avere un bambino con Sindrome di Down sia notevolmente aumentato, il genitore può anche avere figli con cromosomi normali. La prognosi per l'intelligenza e il rischio di complicanze mediche probabilmente dipende dalla proporzione di cellule anormali (p. es., trisomia 21) in ciascun tessuto diverso, compreso il cervello. Tuttavia, in pratica, questo rischio non può essere previsto con precisione, in quanto non è possibile determinare il cariotipo in ogni singola cellula del corpo. Alcune persone con mosaicismo della Sindrome di Down hanno segni clinici molto sottili e possono avere un'intelligenza normale, sebbene anche le persone con Sindrome di Down senza mosaicismo possano presentare reperti clinici variabili.
Fattori di Rischio: L'Età Materna e la Sua Influenza
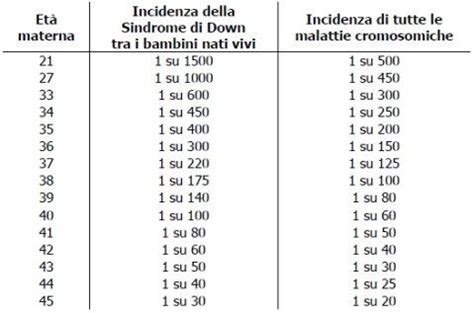
La trisomia 21 è strettamente legata all’età materna: con l’aumento di questa, infatti, aumenta anche il rischio del concepimento di un figlio affetto da Sindrome di Down. L'età materna avanzata è un fattore di rischio riconosciuto per l'insorgenza della Sindrome di Down. Ad esempio, a 20 anni il rischio è di 1 su 1445, mentre a 35 anni, le probabilità previste sono di 1/352 nascite; a 40 anni, sono di 1/85; e a 45 anni è 1 su 25.
Capitolo 3: Il Quadro Clinico: Manifestazioni e Sintomi della Sindrome di Down
La Sindrome di Down, o Trisomia 21, si manifesta attraverso un fenotipo variabile che colpisce molteplici sistemi corporei, causando sia difetti strutturali che funzionali. Non tutte le manifestazioni sono presenti in ogni persona affetta, e il grado di severità può variare ampiamente, rendendo ogni individuo unico. I tratti somatici di un soggetto affetto da Sindrome di Down caratterizzano una facies tipica, ma è importante ricordare che ogni persona è unica e può presentare tali caratteristiche in misura variabile o non presentarle affatto.
Caratteristiche Fisiche Distintive: Dalla Facies ai Tratti delle Mani e dei Piedi
Aspetto generale alla nascita: I neonati affetti tendono a essere tranquilli, piangono raramente e presentano ipotonia muscolare, ovvero un basso tono muscolare. La maggior parte ha un profilo facciale piatto, soprattutto un appiattimento del ponte nasale. Tuttavia, alcuni neonati potrebbero non avere evidenti caratteristiche fisiche inusuali alla nascita e sviluppare successivamente durante l'infanzia le caratteristiche facciali più evidenti. Sono frequenti anche un occipite appiattito, microcefalia (capo più piccolo) ed eccesso di cute nella parte posteriore del collo, che si configura come un collo spesso e tozzo con una plica laterale (pterigio).
Caratteristiche cranio-facciali: La facies tipica comprende una radice nasale appiattita, mento piccolo, macroglossia (lingua grossa e protrusa, spesso rugosa e che può non avere una scissura centrale), e padiglioni auricolari piccoli e a impianto basso. Gli occhi presentano un'angolazione diretta verso l'alto sul bordo laterale (upslanting delle fessure palpebrali, oblique dal basso verso l’alto e dall’interno verso l’esterno) e di solito sono presenti pieghe epicantali agli angoli interni. Potrebbero essere visibili anche le macchie di Brushfield, ovvero piccole macchie grigie o bianche simili a grani di sale, situate perifericamente rispetto all'iride. La bocca è spesso mantenuta aperta, e il palato può essere ogivale e stretto. Le orecchie sono spesso piccole e arrotondate.
Anomalie delle estremità: Le mani sono usualmente piccole e larghe, con dita brevi e tozze. È di frequente rilievo una plica palmare unica trasversale, spesso bilaterale. Il quinto dito può presentare clinodattilia (incurvamento) e spesso ha solo due falangi. Il piede può presentare un'ampia distanza fra il primo e il secondo dito (dita a sandalo), e il solco plantare spesso si prolunga posteriormente sul piede.
Deficit Intellettivo e Sviluppo Psicomotorio
La maggior parte delle persone affette presenta un grado di deterioramento cognitivo, che varia da grave (QI 20-35) a lieve (QI 50-75). Il QI medio è circa 50, ma questo varia ampiamente tra gli individui. Ritardi motori e del linguaggio sono evidenti fin dalla giovane età. Quando i bambini affetti crescono, il ritardo fisico e lo sviluppo mentale diventano più evidenti. Spesso, durante l'infanzia, il comportamento suggerisce disturbi di attenzione e iperattività, e l'incidenza del comportamento associato al disturbo dello spettro autistico risulta aumentata, in particolare nei bambini con grave disabilità intellettiva. È distintivo il fatto che nei soggetti con Sindrome di Down i progressi che si ottengono sul piano dell’apprendimento sono instabili, nel senso che possono essere facilmente perduti. Vi è anche un aumentato rischio di depressione nei bambini e negli adulti con Sindrome di Down.
Comorbidità e Problemi di Salute Associati
La Sindrome di Down comporta vari rischi in termini di salute, con una frequenza superiore rispetto alla popolazione generale. È importante una valutazione clinica periodica per la prevenzione e la diagnosi precoce di alcune patologie, nonché l’individuazione e il trattamento delle problematiche psicomotorie relate alla patologia.
Apparato cardiovascolare: le sfide del cuore: Circa il 40-60% dei bambini (o circa il 50% dei neonati) affetti è associata una cardiopatia congenita. I difetti del setto interventricolare e i difetti del setto atrioventricolare (chiamati anche difetto dei cuscinetti endocardici o difetti del canale atrioventricolare) sono i più frequenti. Altri difetti comuni includono il Botallo pervio, spesso DIA (difetto interatriale) e meno frequentemente DIV (difetto interventricolare). I neonati con difetti cardiaci congeniti possono essere asintomatici o mostrare segni di insufficienza cardiaca, come respiro affannoso, aumentata frequenza respiratoria, difficoltà di alimentazione, sudorazione e scarso aumento di peso. Soffi possono non essere sempre presenti, tuttavia, sono possibili un certo numero di soffi differenti. Alcuni difetti si risolvono spontaneamente (Botallo pervio, spesso DIA, meno frequentemente DIV) necessitando quindi unicamente un monitoraggio clinico/ecocardiografico. Gli altri devono essere corretti chirurgicamente entro i primi anni di vita in relazione alla compromissione clinica.

Sistema gastrointestinale: problematiche digestive comuni: Circa il 6% delle persone affette ha anomalie gastrointestinali, in particolare atresia duodenale, talvolta con pancreas anulare. Possono essere frequentemente riscontrate altre malformazioni congenite a carico dell'apparato gastroenterico, come la malattia di Hirschsprung e l'ano imperforato. I neonati con malattia di Hirschsprung di solito hanno un ritardo nell'evacuazione del meconio. I neonati gravemente colpiti possono avere segni di occlusione intestinale, come vomito biliare, mancata progressione di feci e distensione addominale. Atresia o stenosi duodenale possono manifestarsi con vomito biliare o senza sintomi, a seconda dell'entità della stenosi; questi difetti possono essere rilevati mediante l'ecografia prenatale, mostrando il cosiddetto "segno della doppia bolla".
Sistema endocrino e metabolismo: tiroide, diabete e obesità: Molte persone sviluppano endocrinopatie. Sono frequenti patologie autoimmuni, in particolare distiroidismi (il più delle volte ipotiroidismo), diabete mellito e celiachia, e patologie metaboliche come l'obesità e il diabete mellito di tipo 2. Circa un individuo su due è sovrappeso od obeso. Questo può essere dovuto al metabolismo inferiore alla norma, al basso tono muscolare (ipotonia) e ad eventuali anomalie endocrine (come l’ipotiroidismo), ma anche ad una non adeguata alimentazione e a scarso esercizio fisico.
Occhi e orecchie: prevenzione e diagnosi precoce: Problematiche oculistiche sono quasi generalizzate, interessando circa il 60% delle persone. Tra queste, strabismo e vizi di rifrazione (come la miopia) vanno diagnosticati e corretti precocemente per evitare difficoltà aggiuntive, mentre durante l’adolescenza va effettuato lo screening per il cheratocono. Altre problematiche oculari includono cataratte congenite e glaucoma. La maggior parte delle persone ha una perdita dell'udito, e le infezioni dell'orecchio sono molto frequenti, spesso causate dalle alterazioni morfo-strutturali tipiche a carico del massiccio facciale. La prevenzione dei problemi uditivi nel bambino è fondamentale per evitare difficoltà aggiuntive all’acquisizione di una buona capacità di comunicazione. Anche anomalie a livello odontoiatrico sono comuni, in particolare sovraffollamento dentario o l’assenza di alcuni elementi dentari e palato stretto. Molta attenzione deve essere dedicata, fin dai primi anni di vita, al controllo regolare della vista, dell'udito e dei denti.
Sistema muscolo-scheletrico e neurologico: ipotonia e rischi neurologici: Ipotonia muscolare e lassità articolare sono pressoché costanti. La statura è spesso ridotta, e vi è un deficit staturale; per tale motivo sono state ideate delle curve di crescita apposite per bambini e adolescenti affetti dalla Sindrome di Down. L'ipermobilità atlanto-occipitale e atlanto-assiale, così come anomalie ossee della colonna cervicale, possono causare instabilità atlanto-occipitale e cervicale, con la possibilità di insorgenza di debolezza e paralisi. Una caratteristica neurologica peculiare è la degenerazione del sistema nervoso, che configura un quadro clinico simile alla malattia di Alzheimer, con un precoce invecchiamento e un rischio di demenza più alto rispetto alla popolazione normale. Manifestazioni cliniche potenzialmente associate includono anche sindromi epilettiche e l'apnea durante il sonno.
Sistema immunitario e suscettibilità alle infezioni: Ciò che avviene più frequentemente è la riduzione dei linfociti, associata anche ad un titolo anticorpale inferiore al normale. Le persone con Sindrome di Down hanno una maggiore sensibilità alle infezioni, in particolare quelle delle vie aeree e dell’apparato uditivo. La sindrome aumenta anche la suscettibilità a determinati tipi di cancro, come alcune forme di leucemia durante l’infanzia.
Rischi a lungo termine: invecchiamento precoce e demenza: Oltre all'invecchiamento precoce, problemi di salute nelle persone Down in età adulta comprendono, in ordine crescente, anomalie cardiache congenite e acquisite (30%), malattie polmonari croniche (30%), epilessia (37%), demenza presenile tipo Alzheimer (42%), osteoporosi con conseguente frattura delle ossa lunghe (50%), deficit sensoriali acquisiti (50%) e problemi comportamentali (50%), con una perdita delle abilità cognitive stimata tra il 55% e il 75%.
Capitolo 4: La Diagnosi della Sindrome di Down: Dallo Screening Prenatale alla Conferma Postnatale
La diagnosi della Sindrome di Down può essere effettuata sia nel periodo prenatale, attraverso un programma di screening e successivi test diagnostici di conferma, sia alla nascita, basandosi sul fenotipo caratteristico e confermata da indagini citogenetiche. Lo screening e la diagnosi prenatale devono essere offerti a tutte le persone in gravidanza con informazioni chiare su benefici, limiti e alternative. Il consenso informato e la decisione condivisa sono pilastri fondamentali, così come la tutela dei dati genetici (categoria speciale ai sensi del GDPR).
Screening Prenatale
Lo screening prenatale mira a stimare il rischio che il feto sia affetto da anomalie cromosomiche, inclusa la Sindrome di Down. Diverse metodologie, sia non invasive che invasive, sono disponibili.
Test non invasivi: ecografia fetale e marcatori biochimici:L'ecografia fetale e i test sierici materni sono offerti a tutte le donne incinte in contesti con elevate risorse. L'ecografia fetale può rilevare anomalie come un aumento della translucenza nucale (NT), un difetto del setto atrioventricolare e un'atresia duodenale. Tuttavia, è importante notare che questi segni potrebbero non essere presenti in tutti i feti affetti da trisomia 21. Nel primo trimestre, l’aumento dell’NT e l’assenza o l’ipoplasia dell’osso nasale aumentano il sospetto di Trisomia 21.
I dosaggi sul siero materno possono mostrare livelli anormali di proteina plasmatica A (PAPP-A) alla fine del primo trimestre e alfa-fetoproteina, beta-hCG (gonadotropina corionica umana) libera, estriolo non coniugato e inibina nei primi mesi del secondo trimestre (tra la 15a e la 16a settimana di gestazione). Alcuni markers biochimici sono in grado di valutare il rischio di affezione anche durante il secondo semestre.
Il test combinato:Generalmente, tra 11+0 e 13+6 settimane, quando la CRL (lunghezza cranio-sacrale) è compresa fra 45 e 84 mm, si esegue l’ecografia con misurazione della translucenza nucale (NT) e la valutazione di altri marker (osso nasale, flusso nel dotto venoso, rigurgito tricuspide ove previsto dai protocolli), abbinata ai marcatori biochimici materni PAPP-A e hCG libera. Il “test combinato” offre una buona performance di screening per Trisomia 21 e altre aneuploidie, con valori riportati in letteratura attorno all’82-90% di rilevazione a falsi positivi del 3-5% in contesti controllati. Se il valore di probabilità è compreso tra 1 e 1/250, c’è un’alta probabilità che il feto sia affetto da trisomia 21.
Screening prenatale non invasivo (NIPT/cfDNA): precisione e limiti:Dalla decima settimana è disponibile lo screening non invasivo su sangue materno, basato sull’analisi del cfDNA (cell-free DNA) placentare. Per la Trisomia 21, la sensibilità e specificità di questo test sono molto elevate (≈ >99% e >99% rispettivamente in numerosi studi), mentre il valore predittivo positivo varia con il rischio pre-test (età materna, storia, esito di altri screening). Il cfDNA resta uno screening: un risultato “ad alto rischio” non è una diagnosi e va confermato con test diagnostico invasivo prima di prendere decisioni cliniche importanti.Fattori che possono ridurre la "fetal fraction" (la quantità di DNA fetale nel sangue materno) includono un BMI elevato, un prelievo troppo precoce, patologie autoimmuni e, in alcuni studi, la terapia anticoagulante (eparina), che possono aumentare il tasso di risultati indeterminati ("no-call"). Il cfDNA riflette il genoma placentare, quindi mosaicismo placentare confinato, mosaicismo materno o, raramente, condizioni materne (p. es. neoplasie) possono spiegare risultati cfDNA discordanti con il cariotipo fetale. Alcuni test cfDNA offrono l’analisi “genome-wide” di rare trisomie autosomiche e microdelezioni. Lo screening negativo non azzera il rischio, ma lo riduce drasticamente. In caso di gravidanza gemellare, l'interpretazione del cfDNA è più complessa.
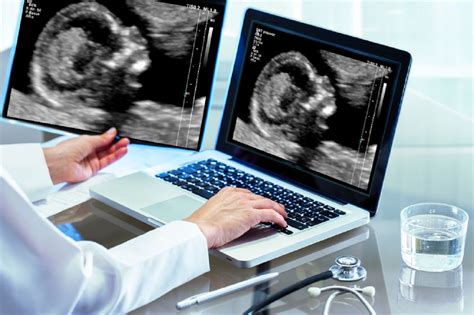
Ecografia morfologica: un controllo essenziale:L’ecografia morfologica, eseguita tra 18 e 22 settimane, è raccomandata per tutte le gravidanze. Nel secondo trimestre, i “soft marker” isolati (p. es. cisti plessochoroidei, focolaio iperecogeno intracardiaco, peli ureterali dilatati, intestino iperecogeno) hanno un valore aggiunto limitato se lo screening è negativo; l’attenzione si concentra su malformazioni maggiori (es. cardiopatie, anomalie gastrointestinali).
Diagnosi Prenatale Invasiva
Se la Sindrome di Down è sospettata sulla base di test di screening (siero materno, ecografia o screening prenatale non invasivo), si raccomanda un test di conferma fetale. Le procedure invasive permettono di ottenere campioni di cellule fetali per l'analisi cromosomica diretta. La diagnosi prenatale, con amniocentesi o prelievo dei villi coriali, dovrebbe essere proposta a tutte le donne gravide con 35 anni o più, in quanto considerate maggiormente a rischio. Le prestazioni per la tutela della maternità rientrano nei LEA (Livelli Essenziali di Assistenza) e sono in esenzione ticket secondo calendari e condizioni definite, con codici “M” indicati sulle ricette dematerializzate.
Villocentesi e amniocentesi: quando e perché:La villocentesi si esegue in genere tra 10 e 13 settimane e campiona i villi coriali placentari. L’amniocentesi si pratica dal secondo trimestre (≥15 settimane), tipicamente tra la 16° e la 18° settimana, e campiona il liquido amniotico. Entrambe consentono diagnosi citogenetica o genomica del feto. Nelle coorti moderne con operatori esperti, il rischio aggiuntivo di perdita fetale associato a queste procedure è basso (nell'ordine di pochi per mille).
Analisi citogenetiche: cariotipo, FISH, microarray:L'indagine citogenetica standard, eseguibile su linfociti periferici, è il test di scelta per la conferma della diagnosi. L'analisi del cariotipo permette di visualizzare i cromosomi e confermare la presenza del cromosoma 21 extra. La conferma può essere fatta anche con FISH (Fluorescence In Situ Hybridization) e con l'analisi cromosomica con microarray. In presenza di malformazioni fetali all’ecografia, il microarray è raccomandato perché aumenta il tasso diagnostico rispetto al cariotipo standard.
Cordocentesi: un'opzione per casi specifici:Il prelievo percutaneo di sangue ombelicale (cordocentesi) è una procedura invasiva utilizzata per ottenere sangue fetale direttamente dal cordone ombelicale, consentendo un cariotipo rapido e la conferma della Trisomia 21. Questa procedura è in genere riservata a situazioni in cui altri metodi diagnostici non sono conclusivi o quando sono necessari risultati rapidi.
Screening in gravidanza e diagnosi prenatale - Dr.ssa Claudia Tironi
Diagnosi alla Nascita: Sospetto Clinico e Conferma Genetica
Il sospetto di Sindrome di Down può essere posto alla nascita sulla base del fenotipo caratteristico del neonato, tra cui la forma del capo o il peso e le dimensioni ridotte rispetto alla media. Tuttavia, per una diagnosi definitiva, è necessaria una conferma genetica. Questa avviene attraverso un test genetico a partire dal sangue del neonato, che permette di definire in modo inequivocabile la presenza della copia extra di cromosoma 21 nelle cellule tramite l'indagine citogenetica del cariotipo.
Rischi di Ricorrenza in Gravidanze Successive
Dopo una gravidanza con trisomia 21 “libera”, il rischio in una successiva gravidanza è circa l'1% oltre al rischio legato all’età materna (o il maggiore fra i due). Se la trisomia 21 è dovuta a traslocazione Robertsoniana, è indicato il cariotipo dei genitori. Con genitori normali (non portatori), il rischio residuo è in genere di circa il 2-3%. Se un genitore è portatore, il rischio dipende dal sesso del portatore e dal cromosoma coinvolto: una madre portatrice ha un rischio del 10-15%, mentre un padre portatore ha un rischio inferiore, circa l'1-3%.
Capitolo 5: Gestione, Assistenza e Qualità della Vita
La Sindrome di Down è una condizione permanente, che può essere curata nelle sue manifestazioni ma non guarita. La terapia consiste nel follow-up e nel trattamento delle singole manifestazioni cliniche, con l'obiettivo di garantire alla persona con Sindrome di Down il raggiungimento del massimo grado possibile di autonomia e una vita piena e soddisfacente.
La Valutazione Clinica Periodica e il Follow-up
Nei pazienti affetti è importante una valutazione clinica periodica per la prevenzione e la diagnosi precoce di alcune patologie, che nella Sindrome di Down si presentano con frequenza superiore rispetto alla popolazione generale, nonché l’individuazione e il trattamento delle problematiche psicomotorie correlate alla patologia. Soprattutto nei primi anni di vita, l’aiuto di un pediatra è essenziale sia per la cura generale che per coordinare le visite specialistiche necessarie. Questo approccio multidisciplinare è fondamentale per gestire la complessità delle comorbidità che possono accompagnare la sindrome.
Trattamento delle Singole Manifestazioni Cliniche
La gestione della Sindrome di Down richiede un'attenzione specifica e individualizzata per ogni singola manifestazione clinica:
- Cardiopatie congenite: Molti difetti cardiaci, come il Botallo pervio o alcuni difetti del setto, possono risolversi spontaneamente, richiedendo unicamente un monitoraggio clinico ed ecocardiografico. Altri difetti, più complessi, devono essere corretti chirurgicamente entro i primi anni di vita in relazione alla compromissione clinica del paziente.
- Problemi oculistici: Strabismo e vizi di rifrazione vanno diagnosticati e corretti precocemente, mentre durante l’adolescenza va effettuato lo screening per il cheratocono. Controlli regolari della vista sono essenziali.
- Problemi uditivi: La prevenzione dei problemi uditivi nel bambino è fondamentale per evitare difficoltà aggiuntive all’acquisizione di una buona capacità di comunicazione, attraverso screening audiometrici regolari e la gestione delle infezioni dell'orecchio.
- Patologie autoimmuni ed endocrine: Devono essere monitorati e trattati distiroidismi, diabete mellito e celiachia, con controlli periodici del funzionamento tiroideo e screening per il diabete.
- Gestione del peso: Data la frequenza di obesità e sovrappeso, è cruciale adottare un'alimentazione adeguata e promuovere l'esercizio fisico per contrastare il metabolismo inferiore alla norma e il basso tono muscolare.
- Sviluppo psicomotorio e cognitivo: L'intervento precoce, attraverso terapie riabilitative e programmi educativi personalizzati, è fondamentale per supportare lo sviluppo cognitivo e motorio, massimizzando il potenziale di apprendimento e autonomia.
L'Importanza della Prevenzione e dell'Intervento Precoce
La prevenzione, intesa in senso ampio, comprende tutto ciò che può essere fatto per evitare conseguenze negative: deficit, disabilità, svantaggi, ma anche diminuzione della salute fisica, del funzionamento motorio, cognitivo, emotivo, affettivo e sociale. Per questo motivo, è fondamentale che le persone con Sindrome di Down seguano abitudini e comportamenti salutari e si sottopongano a controlli puntuali, di modo che i segni e sintomi precoci di complicanze possano essere colti tempestivamente e gli interventi e l’assistenza medica possano essere immediati e personalizzati. Questo approccio proattivo migliora notevolmente la qualità di vita e l'aspettativa.
Aspettativa di Vita: Un Progresso Significativo
Rispetto al passato, le aspettative di vita delle persone con Sindrome di Down sono decisamente aumentate. In meno di cento anni, si è passati da una media di 10 anni a circa 60 anni. Si stima che nei Paesi industrializzati l'aspettativa di vita media oggi superi i 55-60 anni, con variabilità legata alle comorbidità e al miglioramento dell’assistenza sanitaria. Negli anni Sessanta, era di appena 15 anni. Tale aumento è dovuto a un miglioramento della qualità della vita a tutti i livelli, inclusi i progressi nella cura delle complicanze mediche, una maggiore inclusione sociale e un'assistenza sanitaria più mirata e specializzata.
L'Inclusione Sociale e il Potenziale delle Persone con Sindrome di Down
Ci sono circa 38.000 persone affette da Sindrome di Down in Italia. Studiano, lavorano, si muovono e vivono in mezzo a noi, hanno amici, conducono insomma una vita in tutto e per tutto analoga alla nostra. Questo perché si tratta di persone capaci di fare molto di più di quello che siamo abituati a pensare. Esistono da tempo numerose occasioni di sensibilizzazione sulla Sindrome di Down, in cui si cerca di affrontarne diversi aspetti, cercando di chiarire i dubbi e aumentare le conoscenze.
L’obiettivo è favorire, per le persone con la sindrome di Down, una maggiore inclusione nella società. Spesso però non trovano le condizioni adatte per mettere a frutto il proprio potenziale, e anzi spesso incontrano barriere che ne ostacolano la partecipazione alla vita sociale. Tra le ragioni vi è spesso una scarsa consapevolezza, da parte di molte persone, di che cosa si intenda per inclusione. Inoltre, molti sanno poco o nulla di questa specifica forma di disabilità e di cosa comporti per le persone colpite. Il servizio Sanitario Nazionale (SSN) offre prestazioni per la tutela della maternità rientranti nei LEA, e l'offerta del NIPT tramite SSN varia per Regione (universalità, criteri di accesso, fasce di rischio o progetti pilota), altrimenti è disponibile in intramoenia o privato. Promuovere una cultura dell'inclusione significa riconoscere e valorizzare le capacità e i contributi che ogni individuo, incluse le persone con Sindrome di Down, può offrire alla società.
Capitolo 6: Orizzonti Futuri: La Ricerca sulla Sindrome di Down
La ricerca sulla Sindrome di Down è attiva su molti fronti, con un impegno crescente volto a migliorare la comprensione, la diagnosi e la gestione di questa condizione genetica. Si studiano per esempio i meccanismi molecolari con cui l’anomalia genetica esercita i suoi effetti, cercando di identificare quali geni sul cromosoma 21 in eccesso siano i principali responsabili delle diverse manifestazioni cliniche. L'obiettivo è comprendere come la triplicazione genica influenzi lo sviluppo cerebrale, il sistema immunitario, il metabolismo e la suscettibilità a specifiche patologie.
Inoltre, i ricercatori cercano metodi per rendere la diagnosi più precisa, non solo nello screening prenatale ma anche nella caratterizzazione delle varianti della sindrome. Si approfondiscono i meccanismi patologici alla base delle comorbidità, come le cardiopatie congenite, i disturbi tiroidei, la malattia di Alzheimer a esordio precoce e l'aumentato rischio di leucemia. Questa comprensione dettagliata è fondamentale per sviluppare nuovi trattamenti mirati per queste complicanze, migliorando la qualità e l'aspettativa di vita.

Un obiettivo a lungo termine, che per ora è soprattutto un sogno, è quello di sviluppare una vera e propria cura per la Sindrome di Down. Questi ambiziosi obiettivi sono perseguiti studiando l'anomalia genetica e i suoi possibili rimedi in animali di laboratorio, come modelli murini della trisomia 21. Attraverso questi studi, i ricercatori esaminano gli effetti sia di geni specifici che di gruppi di geni che possono essere coinvolti nella sindrome, cercando potenziali target terapeutici che potrebbero mitigare gli effetti della triplicazione cromosomica. I progressi nella genetica e nelle neuroscienze offrono nuove speranze per interventi futuri, che potrebbero un giorno andare oltre la gestione sintomatica per affrontare la causa sottostante della Sindrome di Down.
tags: #diagnosi #trisomia #21 #libera #esperienze