Nei neonati, la craniostenosi è spesso individuata attraverso il monitoraggio della crescita della testa e la forma cranica. La craniostenosi si caratterizza principalmente per la fusione prematura di una o più suture craniche, che porta a deformità del cranio. Un bambino nei primi mesi di vita può presentare un'asimmetria della testa, e questa anomalia può essere facilmente apprezzabile. L’aspetto del volto del bambino spesso non è simmetrico. Questa condizione può essere osservata o, meno comunemente, la sinostosi può causare un aumento della pressione intracranica, in particolare quando le suture prematuramente fuse sono multiple. I cambiamenti di forma della testa e del viso sono facilmente apprezzabili e possono essere il primo e unico sintomo. Un altro segno sono le fontanelle piccole o assenti.
È fondamentale che la patologia venga correttamente diagnosticata ed i bambini possano essere sottoposti al corretto trattamento chirurgico entro i primi anni di vita, quando questo permette di ottenere i migliori risultati sia da un punto di vista funzionale sia estetico.
Comprendere la Craniostenosi: Il Meccanismo Sottostante
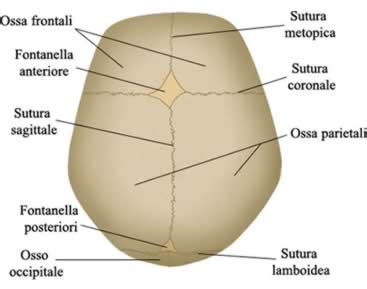
Il cranio normale è costituito da una serie di ossa piatte separate da suture. Le suture (giunzioni fibrose) si trovano tra le ossa piatte della testa. La craniostenosi è una condizione in cui le suture si fondono troppo precocemente, causando problemi al normale sviluppo parenchimale e cranico. Questo deriva dal greco e significa chiusura, ristrettezza del cranio. La Craniostenosi è una delle malformazioni congenite più frequenti, per la quale viene riportata un’incidenza di circa 1 neonato su 2000. La malformazione è caratterizzata dalla prematura saldatura di una o più suture craniche, che comporta una crescita disarmonica del cranio durante i primi due anni di vita.
In condizioni normali, il cervello dei neonati si sviluppa aumentando il proprio volume in armonia con la progressiva crescita ossea del cranio, in un processo che si conclude, di solito, entro i due anni. Alla nascita, infatti, le ossa del cranio sono separate dal tessuto fibro-cartilagineo delle suture e dalle fontanelle, che sono zone fibrose che si trovano all’incrocio delle suture. In condizioni di normalità, la fusione delle suture craniche avviene in epoca post-natale e termina, per alcune ossa del cranio, alla soglia dei 20 anni. Il cranio è infatti composto da ossa piatte, separate da suture, che si chiudono normalmente fra il primo ed il terzo anno di vita. Man mano che il cervello dei bambini si sviluppa nei primi due anni di vita, raddoppiando il suo volume, le suture permettono al cranio di espandersi progressivamente e grazie a questa “spinta modellante” si ottiene la crescita armonica della testa.
Se una o più di queste suture si chiude prematuramente - come accade nel caso delle craniosinostosi - il cranio si espande lungo le suture che rimangono aperte causando una malformazione. Quando una sutura si chiude e le ossa del cranio si uniscono prematuramente, la testa smette di crescere solo su quella parte del cranio. Dove le suture non si sono unite, la testa del bambino continua a crescere. Quando ciò accade, il cranio avrà una forma anormale, anche se il cervello all’interno ha raggiunto le sue dimensioni normali. Quando una o più suture sono chiuse, il cranio si espande lungo le suture che rimangono aperte, con una conseguente progressiva deformazione della sua struttura, che comporta una conformazione disarmonica ed anomala del cranio.
Le Diverse Manifestazioni della Craniostenosi: Una Classificazione Dettagliata
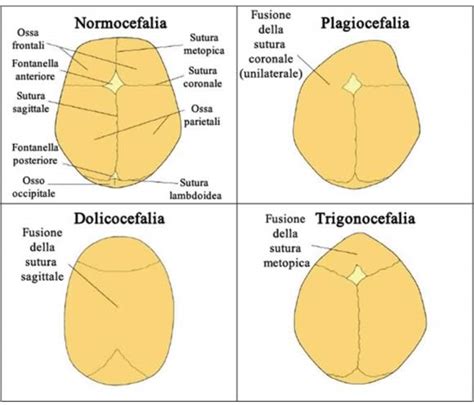
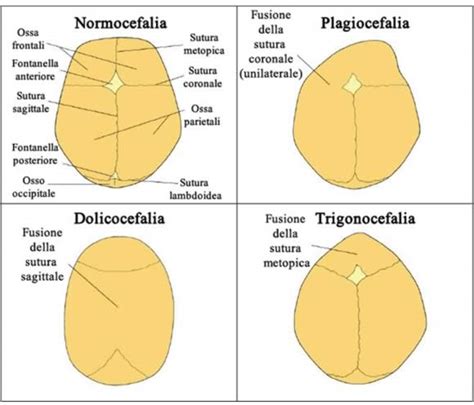
Esistono fondamentalmente 4 tipi di craniosinostosi, in base alla sutura che si chiude prematuramente. I sintomi comuni includono una forma irregolare del cranio, che varia a seconda della sutura interessata. Le diverse craniostenosi si presentano in modo stereotipato con morfologie tra loro differenti in base alla sutura/alle suture affetta/e.
Le craniosinostosi più comuni nei neonati includono:
- Scafocefalia (o Dolicocefalia): È la più comune craniosinostosi con una prevalenza di circa 1:5.000 bambini nati. La scafocefalia è una fusione precoce della sutura sagittale. La sutura decorre da davanti a dietro lungo la linea sagittale. Una fusione precoce di questa sutura causa una forma allungata e stretta del cranio (testa lunga per il tipico aumento del diametro antero-posteriore) o più comunemente scafocefalia (= la conformazione dello scafo della nave). La fusione della sutura sagittale può causare una testa allungata e stretta. La predominanza è nel sesso maschile.
- Plagiocefalia Anteriore (o Sinostosi Coronale Unilaterale): Questa condizione è anche nota come sinostosi coronale e blocca il normale sviluppo della fronte e dell’arcata sopraccigliare. A volte può causare un appiattimento della fronte e dell’arcata sopraccigliare solo del lato affetto con una tendenza a una eccessiva prominenza della fronte contro lateralmente. L’occhio dal lato affetto potrebbe anche avere una forma differente. Il coinvolgimento di una sola sutura coronarica avviene nella metà dei casi ed è definita plagiocefalia anteriore (sinistra o destra a seconda del lato in cui si è verificata la saldatura). Ha la prevalenza di circa 1:10.000 bambini nati e la maggioranza dei casi sono sporadici, con una predominanza per il sesso femminile (rapporto M.F = 1:2). La fusione di una o entrambe le suture coronali porta ad una conformazione del cranio sviluppata in verticale con asimmetria della fronte e delle orbite.
- Brachicefalia (o Sinostosi Coronale Bilaterale): Il coinvolgimento bilaterale della sutura coronale dà origine ad una conformazione cranica corta, definita brachicefalia con riduzione del diametro anteroposteriore del cranio. Questa è la sinostosi coronale bilaterale, che può portare a una turricefalia.
- Trigonocefalia (o Sinostosi Metopica): La trigonocefalia è la fusione della sutura metopica (frontale). Questa sutura decorre dalla sommità della testa lungo la metà della fronte, verso il naso. Una saldatura precoce di questa sutura potrebbe produrre una cresta prominente lungo la fronte. La fusione della sutura metopica avviene nel periodo gestazionale e dà origine alla cosiddetta trigonocefalia; ha una prevalenza alla nascita di circa 1:15.000 con una predominanza nel sesso maschile (M:F=3:1). Circa il 5-6% dei casi sono famigliari.
- Plagiocefalia Posteriore (o Sinostosi Lambdoidea): La plagiocefalia posteriore o sinostosi lambdoidea presenta una prevalenza di 1:15.000 bambini nati con una predominanza nel sesso maschile. Questa si estende lungo la parte posteriore della testa. Si caratterizza per un appiattimento della regione parieto-occipitale e dislocazione verso il basso della regione mastoidea omolaterali con conseguente patologica inclinazione del basicranio, espansione compensatoria parietale controlaterale.
- Oxicefalia: La chiusura di più suture, che comporta una crescita “puntuta” del cranio (verso la fontanella anteriore, che chiude per ultima) viene definita oxicefalia.
- Pachicefalia: La chiusura delle due suture lambdoidee comporta una piattezza dell’occipite e viene chiamata pachicefalia.
Le sinostosi complesse o polisuturali (definite come la contemporanea chiusura di più di una sutura) hanno un’incidenza di circa il 5% dei casi non sindromici (cioè non associata a definiti quadri clinici complessi causati da alterazioni genetiche note). Il coinvolgimento di due suture avviene in circa i 2/3 dei casi, e l’interessamento di due o più suture in un terzo dei casi. Spesso non interessano suture contigue. La sinostosi coinvolge, più frequentemente, una singola sutura (forma monosuturale o semplice), solitamente “maggiore”: la metopica, la sagittale, l’emicoronale destra o sinistra, di rado la lambdoidea destra o sinistra. È possibile, inoltre, la sinostosi contemporanea di più suture (forma multisuturale), condizione spesso riconducibile a quadri malformativi più complessi (spesso sindromici). Non infrequente è il coinvolgimento della regione orbitaria, mono o bilaterale, più raro quello della regione centrofacciale.
Sintomi e Riconoscimento Precoce della Craniostenosi
I sintomi nei neonati possono essere meno evidenti e talvolta sono scoperti solo attraverso esami di routine o per altre condizioni mediche. Oltre alle deformità della testa, i segni visibili possono includere una chiusura precoce della fontanella anteriore, e una crescita anomala della testa. La simmetria del viso e del cranio può essere compromessa, con asimmetrie evidenti nelle orbite oculari o nelle orecchie. Questi cambiamenti nella forma della testa e del viso possono essere evidenti.
Sintomatologicamente le craniostenosi vanno classificate in compensate e scompensate.
- Forme compensate: Presentano un accrescimento del cervello normale senza apparenti segni di compressione del sistema nervoso. I segni clinici più evidenti riguardano principalmente lo scheletro, deformazione cranica, chiusura precoce della fontanella bregmatica, ridotto perimetro cranico e alterazioni facciali.
- Forme scompensate: Sono palesemente più gravi, e determinano una sintomatologia d’ipertensione endocranica, che può instaurarsi in modo subdolo e presentare una sintomatologia invalidante.
Nei bambini più grandi e negli adolescenti, i sintomi possono includere mal di testa persistenti, problemi di vista, e difficoltà di apprendimento o di comportamento.
Le Cause della Craniostenosi: Fattori Genetici e Ambientali
La causa delle Craniostenosi è ancora sconosciuta; per alcune forme più gravi, che implicano l’interessamento di più suture, è stata riconosciuta una alterazione genetica ed è nota una trasmissione familiare, mentre per quanto riguarda le forme che interessano una sola sutura molta ricerca rimane ancora da fare. Sebbene non siano state ancora identificate le alterazioni genetiche è probabile che ve ne siano, in quanto è osservazione frequente una ricomparsa della malformazione nei figli dei soggetti trattatati.
Le craniostenosi si presentano spesso come anomalie isolate, oppure possono associarsi ad altre malformazioni o condizioni patologiche all’interno di veri e propri quadri sindromici. Si distinguono forme semplici e complesse, in base al tipo e al numero di suture interessate, e forme sindromiche e non sindromiche (isolate), sulla base della presenza o meno di altri segni e sintomi associati (malformazioni in altre regioni del corpo, ritardo dello sviluppo, etc.).
Molteplici sono le classificazioni proposte. Tra queste è utile segnalare la distinzione tra le forme non sindromiche (le più numerose, solitamente monosuturali, frequentemente sporadiche) e le sindromiche (solitamente multisuturali, spesso parte di un disordine sistemico, associate o meno a trasmissione famigliare). Le craniosinostosi semplici (una sola sutura coinvolta) non sindromiche sono le forme più frequenti, pur restando nell’ambito delle malattie rare. Per le varianti isolate è più complicato definire la causa, che spesso può essere multifattoriale, cioè legata ad una combinazione di fattori genetici e ambientali (esposizione ad agenti chimici o fisici durante la gravidanza) non ancora chiariti.
La craniostenosi può essere associata a sindromi genetiche come la sindrome di Apert, Crouzon, o Pfeiffer, che comportano altre malformazioni craniofacciali e anomalie sistemiche. La sindrome di Saethre-Chotzen è un'altra condizione sindromica che può associarsi. La craniostenosi è una caratteristica di molte patologie sindromiche genetiche con differenti pattern ereditari e diverse possibilità di ricorrenza, a seconda della sindrome specifica. È fondamentale, sia per il paziente che per i familiari, che sia esaminato attentamente ogni segno che possa rimandare a una causa sindromica (geneticamente determinata) di craniostenosi. Sono note oltre 90 sindromi con craniostenosi associata di cui circa la metà mostra modalità di trasmissione autosomica dominante o recessiva.
I geni dei recettori dei fattori di crescita dei fibroblasti (FGFRs) e il gene TWIST sono responsabili delle forme comuni di craniostenosi. I recettori dei fattori di crescita dei fibroblasti (FGFRs) sono costituiti da 4 diverse proteine, codificate da 4 geni diversi.Per quanto riguarda le modalità di trasmissione ereditaria, si distinguono:
- Autosomico dominante: Vuol dire che un gene è necessario e sufficiente per esprimere la condizione patologica, e il gene è trasmesso da genitore a figlio con un rischio del 50/50 ad ogni gravidanza.
- Autosomico recessivo: Vuol dire che sono necessarie due copie mutate del gene, ereditate da ognuno dei genitori che sono portatori obbligati. I genitori eterozigoti hanno una chance su quattro, ovvero il 25% di probabilità ad ogni gravidanza di avere un figlio con la craniosinostosi. Maschi e femmine sono ugualmente colpiti.
Sono stati individuati anche dei fattori di rischio da tenere in considerazione a fini diagnostici:
- Una nascita pretermine (meno di 37 settimane),
- Peso inferiore ai 2500gr.,
- Gravidanza gemellare,
- Età paterna oltre i 40 anni (in particolare per la sinostosi coronale),
- Cause iatrogene (sindrome da iperdrenaggio in caso di derivazione liquorale),
- Patologie metaboliche o ematologiche (es. ipertiroidismo, rachitismo, mucopolisaccaridosi, talassemia, anemia falciforme),
- Malformazioni (oloprosencefalia, microcefalia, encefalocele),
- Teratogeni (idantoina, retinoidi).
Gli studi epidemiologici sulle craniostenosi hanno evidenziato una predominanza del sesso maschile, in particolare per le sinostosi sagittale e lambdoidea, mentre per le sinostosi coronali si riscontra una predominanza del sesso femminile. Le anomalie associate sono soprattutto a carico degli arti (84%), anomalie dell’orecchio (38%) e difetti cardiaci congeniti (23%), occorrono più frequentemente nelle sinostosi coronali che nelle sinostosi sagittali.
Il Percorso Diagnostico: Identificare la Craniostenosi per un Intervento Tempestivo
La diagnosi precoce e un trattamento appropriato possono aiutare a prevenire complicazioni a lungo termine e migliorare la qualità della vita dei pazienti. Come afferma la dottoressa Wanda Lattanzi, medico genetista e ricercatore in Biologia Applicata presso l’Università Cattolica del Sacro Cuore a Roma, “è sempre auspicabile un rapido e corretto inquadramento del paziente, alla nascita o nei mesi immediatamente seguenti”.
La diagnosi è solitamente clinica e la sua precocità si riflette positivamente sul perseguimento delle migliori prospettive. Durante la visita medica, il medico dovrebbe ottenere una anamnesi completa prenatale e della nascita, in particolar modo storie familiari di craniosinostosi o altre anomalie craniofacciali. Un altro argomento da approfondire sono le tappe dello sviluppo soprattutto da quando si è sviluppata la craniosinostosi.
Per confermare la craniosinostosi, è necessaria una diagnosi clinica esperta e viene confermata mediante la tomografia computerizzata (TC) del cranio con ricostruzioni tridimensionali. Per pianificare l’intervento chirurgico è utile la pratica di una scansione elicoidale tridimensionale (TC 3D; TC 3D). La Tomografia Computerizzata (chiamata anche TC o TAC) del capo mostra immagini dettagliate di qualunque parte del corpo, incluse ossa, muscoli, grasso e organi. In alcuni centri specializzati è possibile ricorrere ad un esame ecografico ad alta risoluzione che consente di valutare lo stato di ossificazione delle suture. Per un forte sospetto va eseguita una Rx del cranio o meglio una TC. Ci riserviamo l’uso della risonanza magnetica (NMR o MRI) per gli studi sulla massa cerebrale.
“Le comuni tecniche di diagnosi prenatale invasive (amniocentesi, villocentesi) e non (analisi del DNA fetale nel sangue materno) - continua la dottoressa Lattanzi - non consentono di fare diagnosi di mutazioni geniche associate a craniosinostosi. Alcuni studi hanno valutato la possibilità di fare diagnosi prenatale tramite ecografia morfologica ad elevata sensibilità in alcune forme sindromiche, per le quali la gravità delle malformazioni consente un più facile inquadramento ecografico. Questo è meno facile da ottenere per le forme semplici. Tuttavia, il quadro clinico di queste ultime rappresenta una malattia piuttosto 'benigna', per la quale il ricorso a una diagnosi prenatale non risulterebbe indispensabile. Un rapido e corretto inquadramento alla nascita o nei mesi immediatamente seguenti è invece sempre auspicabile."
Craniosynostosis FAQ: How Do You Diagnose Metopic Synostosis?
Le Complicazioni della Craniostenosi Non Trattata
La craniostenosi non trattata può avere un impatto significativo sulla crescita e sullo sviluppo. Nei bambini, può interferire con la normale crescita del cranio e del cervello, portando a complicazioni a lungo termine. La presenza di craniostenosi priva le strutture encefaliche dello spazio che serve a quest'ultime, per crescere in modo corretto. Le complicazioni della craniostenosi possono includere l'idrocefalo, una condizione in cui si accumula liquido cerebrospinale nel cervello, causando ulteriore aumento della pressione intracranica. L'idrocefalo può aggravare i sintomi neurologici e richiedere un trattamento chirurgico per alleviare la pressione.
Nei casi più gravi, nei quali sono interessate più suture, oltre alla deformità del cranio si sviluppa anche una aumentata pressione al suo interno (ipertensione endocranica), che può comportare danni al cervello in evoluzione se non viene correttamente e precocemente diagnosticata e trattata. I danni possono colpire la vista, l’udito e le capacità cognitive. Questa ipertensione intracranica può causare problemi come cecità, convulsioni o danni cerebrali. Le craniosinostosi non sindromiche presentano un’elevata percentuale di ritardi funzionali superiore al 50%.
Per capire l'ipertensione endocranica, si pensi di gonfiare una palla ginnica all'interno di una scatola rigida di ridotte dimensioni: quando la palla raggiunge determinate dimensioni spinge sulle pareti della scatola, esercitando una certa pressione. In condizioni di normalità, le suture craniche non ancora fuse tra loro rendono il cranio modellabile secondo l'accrescimento dell'encefalo. Un bambino con craniosinostosi necessita di frequenti valutazioni mediche al fine di garantire che il cranio, le ossa facciali, e il cervello si sviluppano normalmente.
Il Trattamento Chirurgico: Una Soluzione Necessaria e Tempestiva
Per queste patologie, l’unico trattamento possibile, al momento, è l’intervento chirurgico, affiancato dalle terapie per eventuali sintomi associati. Il trattamento chirurgico è tipicamente il trattamento raccomandato. Il trattamento della malformazione è chirurgico ed ha tre finalità:
- Alleviare l’ipertensione endocranica, prevenendo i danni cognitivi e visivi che questa può comportare.
- Permettere che il cranio accolga e “protegga” la crescita del cervello, coprendolo con una volta ossea.
- Cercare di migliorare l’aspetto di questi bambini da un punto di vista estetico.
Le deformità craniche devono essere trattate il prima possibile (prima dei 6 mesi di età). Il miglior timing per praticare l’intervento chirurgico è prima del 1 anno di età finché l’osso è ancora molto malleabile e le restanti suture non sono ancora completamente fuse e facilmente maneggiabili. Si tende a operare entro il primo anno di vita, per ridurre al minimo i danni da compressione dell’encefalo e delle strutture orbitarie e per ottenere migliori risultati estetici. In alcuni casi la chirurgia potrebbe essere necessaria anche più precocemente a seconda della gravità della condizione. Dato che la perdita ematica potrebbe essere un problema importante di questa chirurgia, si tende a rimandare la data dell’intervento a quando il bambino abbia una quantità sufficiente di volume ematico da poter affrontare la chirurgia. Prima dell’intervento chirurgico proponiamo un check-up oftalmologico, un test di maturazione neurologica e uno studio dismorfologico pediatrico.
Dopo l'operazione, è comune per il bambino di avere una medicazione simile ad un turbante intorno alla testa. Il viso e le palpebre possono essere gonfie dopo questo tipo di intervento chirurgico. Le complicanze postoperatorie possono verificarsi improvvisamente, immediatamente dopo l’intervento o in una maniera differita. Queste complicanze richiedono una pronta valutazione del chirurgo. A prescindere dall'approccio chirurgico utilizzato, l'operazione per l'eliminazione di una craniostenosi prevede sempre, alla sua conclusione, un ricovero ospedaliero della durata di 4-5 giorni. Il trattamento e gestione della Craniostenosi implicano il monitoraggio non chirurgico, la chirurgia per casi gravi e la riabilitazione post-operatoria per una crescita sana.

Distinzione Cruciale: Craniostenosi e Plagiocefalia Posizionale
Quando si parla di deformità craniche, è molto importante differenziare le deformità craniofacciali posizionali dalle craniosinostosi, perché il trattamento e le possibili sequele sono molto differenti. Le craniosinostosi sono dovute alla chiusura prematura di alcune delle più importanti suture craniche che consentono al cranio del bambino di crescere con la crescita del suo cervello.
La plagiocefalia deformativa (posizionale) è una condizione in cui la testa è malformata (asimmetrica) per le ripetute pressioni sulla stessa area della testa. Non si tratta di una vera sinostosi. Le suture sono ancora aperte e normali. La plagiocefalia posizionale è una condizione caratterizzata dalla deformità del cranio, in genere il neonato si presenta con un appiattimento di una porzione cranica mentre un’altra parte presenta un rigonfiamento. Il numero di bambini con plagiocefalia deformativa è aumentata nel corso degli ultimi anni. Questo aumento potrebbe essere il risultato della campagna del “dormire sul dorso” promossa dall’AMERICAN Academy of Pediatrics (AAP) adottata per prevenire le sindromi da morte improvvisa in culla (SIDS), ma anche altri fattori potrebbero causare questo tipo di plagiocefalia.
È importante differenziarla dalla craniostenosi, ovvero un gruppo di malformazioni del cranio, dove una o più suture craniche si sono fuse in maniera precoce. Il trattamento specifico andrà determinato in base alla severità. La plagiocefalia posizionale si tratta con una variazione della posizione usuale di decubito durante il sonno o utilizzando dei caschetti rimodellanti che tentano di rimodellare la forma del cranio con il tempo.
Per quanto riguarda la plagiocefalia posizionale, con una diagnosi tempestiva è possibile correggere l’estetica del cranio tramite una terapia conservativa che comprende sia l’intervento osteopatico e l’introduzione di alcuni accorgimenti a casa da parte dei genitori. Spesso questo problema è associato ad un'altra condizione ovvero il torcicollo miogeno del muscolo sternocleidomastoideo (muscolo che si trova nella zona antero-laterale del collo) che può essere sia di tipo funzionale (causato da uno spasmo o una contrattura senza cambiamenti patologici nel muscolo) o di tipo patologico (causato da una fibrosi del muscolo). Con la continua trazione da parte di questo muscolo della testa, il capo del neonato è costretto a fissare una posizione non idonea, con inclinazione dallo stesso lato del muscolo affetto e rotazione della testa verso il lato opposto. Le conseguenze non sono solo estetiche, questo adattamento può causare dei compensi a livello di tutto il corpo portando all'insorgenza di alcuni dei principali disturbi nel neonato (coliche gassose, reflusso gastro-esofageo o rigurgito e difficoltà nella suzione) ma anche a problematiche di sviluppo motorio, come ritardo nel gattonamento.
Quello che è di primaria importanza per l’andamento del trattamento osteopatico è la tempistica, infatti l’infante dovrebbe essere visitato dal terapista sin dai primi mesi di vita, in modo tale da sfruttare la capacità del cranio di adattarsi a forze esterne. Variare la posizione assunta dal neonato quando è sveglio, promuovendo tutte le posizioni (pancia in su, pancia in giù, sul fianco destro e sinistro) è fondamentale per la correzione.
tags: #craniostenosi #neonato #si #guarisce