La tetralogia di Fallot è una cardiopatia congenita complessa che si sviluppa durante la vita fetale e si manifesta subito dopo la nascita o nei primi mesi di vita. È una malformazione cardiaca congenita che prende il nome dal medico francese Étienne-Louis Arthur Fallot, che la descrisse nel XIX secolo. Questa condizione è caratterizzata dalla combinazione di quattro anomalie strutturali del cuore, che compromettono la normale ossigenazione del sangue e il corretto flusso tra cuore e polmoni. La tetralogia di Fallot rappresenta una delle cardiopatie congenite più comuni, affliggendo circa l'8-10% delle cardiopatie congenite e approssimativamente un neonato su 2500. È la malformazione congenita del cuore più frequente tra quelle che determinano cianosi. In questo contesto, l'aorta a cavaliere rappresenta una delle anomalie centrali, e le sue cause possono includere alterazioni genetiche o sindromi cromosomiche, come la Trisomia 21.
Anatomia e Fisiologia del Cuore
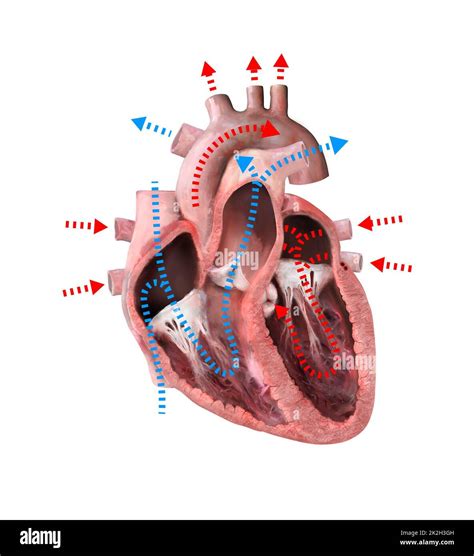
Prima di addentrarsi nella descrizione della tetralogia di Fallot, è utile richiamare alcuni elementi anatomici e funzionali del cuore. In un organismo sano, la funzione del cuore è quella di pompare il sangue prima nei polmoni, dove si ossigena, e poi di inviare il sangue così ossigenato in tutto il corpo. Il cuore è diviso in due metà, destra e sinistra, ognuna delle quali comprende un atrio e un ventricolo. Il cuore di destra è composto dall'atrio destro e dal sottostante ventricolo destro, mentre il cuore di sinistra è composto dall'atrio sinistro e dal sottostante ventricolo sinistro. Ciascun atrio è collegato al ventricolo sottostante per mezzo di una valvola.
Il sangue non ossigenato, proveniente dagli organi e dai tessuti del corpo umano, raggiunge l'atrio destro attraverso le vene cave e viene spinto nel ventricolo destro. Da qui, il ventricolo destro pompa il sangue nell'arteria polmonare, che lo conduce ai polmoni per l'ossigenazione. Nei polmoni, il sangue si carica di ossigeno e rilascia anidride carbonica. Il sangue ossigenato ritorna poi all'atrio sinistro attraverso le vene polmonari, passa nel ventricolo sinistro e da qui viene pompato nell'aorta, la più grande e importante arteria del corpo umano, che distribuisce il sangue ossigenato al resto dell’organismo. Questo ciclo assicura che gli scambi gassosi siano il più efficaci possibile sia a livello polmonare (raccolta di ossigeno e rilascio di anidride carbonica) sia in periferia (rilascio di ossigeno e raccolta di anidride carbonica).
Definizione e Componenti della Tetralogia di Fallot
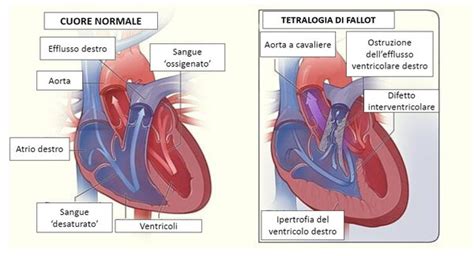
La tetralogia di Fallot è una malformazione del cuore, caratterizzata da quattro anomalie anatomiche presenti contemporaneamente, le cui manifestazioni cliniche dipendono dalla severità del quadro anatomico. Il nome "tetralogia" (dal prefisso "tetra-" che significa quattro) indica la presenza di queste quattro anomalie principali, che alterano il normale flusso di sangue che attraversa il corpo umano e compromettono la normale ossigenazione del sangue.
Le quattro anomalie che costituiscono la tetralogia di Fallot sono:
Stenosi (o ostruzione) dell'efflusso ventricolare destro e/o della valvola polmonare: Si tratta di un restringimento del tratto che collega il ventricolo destro all’arteria polmonare e/o della valvola polmonare stessa. Questa ostruzione può essere più o meno grave e ha l'effetto di ridurre la portata di sangue che imbocca l'arteria polmonare e che, poi, raggiunge i polmoni. Le conseguenze sono un ristagno di sangue nel ventricolo destro e una quota di sangue ossigenato inferiore a quella normale. L'ostruzione può anche interessare le arterie polmonari principali e le sue ramificazioni.
Difetto interventricolare (DIV): È un’apertura anomala, un "buco", nel setto interventricolare, la parete che normalmente separa nettamente i due ventricoli, ovvero le camere inferiori del cuore. Il difetto del setto interventricolare (comunicazione interventricolare) nella tetralogia di Fallot è spesso descritto come in malallineamento, poiché il setto conale è dislocato anteriormente. Il difetto è in genere di grandi dimensioni, il che permette ai due ventricoli di comunicare tra loro. Ciò porta il sangue non ossigenato a travasare dal ventricolo destro e a mescolarsi con il sangue ossigenato del ventricolo sinistro, un fenomeno noto come "shunt da destra a sinistra". Il termine shunt in medicina indica un passaggio che permette il travaso di un liquido da un compartimento a un altro.
Aorta a cavaliere (o malallineamento dell'aorta rispetto al ventricolo sinistro): L’aorta è spostata verso destra rispetto alla sua posizione normale. Invece di ricevere sangue solo dal ventricolo sinistro, riceve sangue da entrambi i ventricoli, a causa anche del difetto del setto interventricolare. Questa origine biventricolare dell'aorta significa che il sangue misto (ossigenato e non ossigenato) imbocca l'aorta e raggiunge i tessuti del corpo umano, contribuendo al colorito cianotico. Questo setto dislocato sporge nel tratto di efflusso polmonare.
Ipertrofia ventricolare destra: A causa della stenosi della valvola polmonare, il ventricolo destro deve lavorare con maggiore forza per spingere il sangue non ossigenato nei polmoni. Per far ciò, il muscolo del ventricolo si accresce e si rinforza, determinando un ispessimento delle pareti cardiache del ventricolo destro. A differenza dello sviluppo muscolare che si osserva ad esempio nel cuore degli atleti, questo tipo di stimolo è legato allo sviluppo di un progressivo irrigidimento delle pareti del cuore, che esitano alla fine in un indebolimento delle stesse. Il risultato finale è che il muscolo del ventricolo perde elasticità e si irrigidisce.
Il risultato finale di questi difetti è l’arrivo di sangue poco ossigenato in aorta e quindi nella circolazione sistemica, che causerà, a seconda della gravità dei difetti, una cianosi più o meno grave. Il risultato finale è che meno sangue verrà ossigenato e che del sangue desaturato prenderà la via dell'aorta attraverso la comunicazione tra i ventricoli. In molti casi si osservano anche difetti cardiaci supplementari, come un difetto interatriale (nel 70% dei casi), ma in ogni caso la conseguenza è che la distribuzione di ossigeno viene gravemente compromessa, fino ad essere insufficiente non appena le richieste aumentano oltre un livello minimo.
Fisiopatologia della Tetralogia di Fallot
La fisiopatologia della tetralogia di Fallot è intrinsecamente legata alle quattro anomalie anatomiche. Il difetto del setto interventricolare (comunicazione interventricolare) è di solito di grandi dimensioni, il che fa sì che le pressioni sistoliche siano uguali nel ventricolo destro e sinistro (e nell'aorta). La fisiopatologia dipende dal grado di ostruzione all'efflusso ventricolare destro. Un'ostruzione di lieve entità può provocare un netto shunt sinistro-destro attraverso il difetto del setto interventricolare; al contrario, un'ostruzione severa causa uno shunt destro-sinistro, determinando bassa saturazione arteriosa sistemica (cianosi) non rispondente a ossigeno supplementare. Il flusso ematico polmonare è diminuito, il ventricolo destro è ipertrofico e sangue non-ossigenato entra nell'aorta. Le pressioni sistoliche nel ventricolo destro, ventricolo sinistro e nell'aorta sono identiche. I livelli della desaturazione arteriosa di ossigeno sono correlati alla gravità dell'ostruzione del tratto di efflusso del ventricolo destro.
Epidemiologia
L'incidenza della tetralogia di Fallot è di 1 caso ogni 3.600 nuovi nati circa. È la malformazione congenita del cuore più comune tra quelle che determinano cianosi. Colpisce in ugual misura uomini e donne.
Cause della Tetralogia di Fallot e Associazione con Trisomia 21
Le cause precise della tetralogia di Fallot, al momento, non sono completamente conosciute. Tuttavia, si ritiene che siano responsabili fattori sia genetici che ambientali. Nell’80-90% dei casi, molteplici fattori contribuiscono allo sviluppo di una cardiopatia congenita, trattandosi di fattori genetici predisponenti e fattori ambientali scatenanti. Questi possono agire singolarmente o, più spesso, combinarsi e agire di concerto. La malformazione insorge in età pre-natale, cioè prima della nascita, durante il periodo di morfogenesi cardiaca, e può coinvolgere solo il cuore, o anche altri organi e apparati.
Cardiopatie congenite: sintomi e cause – Dott. Mario Carminati
Fattori Genetici
La tetralogia di Fallot è associata, molto spesso, ad alcune patologie genetiche e anomalie cromosomiche. Il 10% dei casi è da riferire ad un’anomalia cromosomica. Tra queste, la Sindrome di Down (Trisomia 21) è una delle più rilevanti. La Sindrome di Down è caratterizzata dalla presenza di tre copie del cromosoma 21. Altre sindromi cromosomiche e malattie genetiche associate includono:
- Sindrome di DiGeorge: Caratterizzata da una delezione di una porzione del cromosoma 22.
- Sindrome di Turner: Una monosomia del cromosoma X (solo nelle femmine).
- Sindrome di Williams: Anomalia genetica che può coinvolgere il cromosoma 7.
- Fenilchetonuria materna: Una mutazione puntiforme di alcune basi del DNA nel cromosoma 12.
Il restante 5-10% dei casi è definito monogenico, perché dovuto alla mutazione di un singolo gene, come in alcune sindromi e in alcune cardiopatie isolate in cui è presente una dimostrata trasmissione familiare (uno dei due genitori è affetto). Si è osservata una ricorrenza tra fratelli dell’1-3%.
Fattori Ambientali
Si è osservata una relazione tra alcune circostanze ambientali e lo sviluppo della tetralogia di Fallot. Queste includono:
- Alcolismo materno: Il bambino può ammalarsi di sindrome fetale alcolica.
- Alcune malattie virali della madre: Infezioni che insorgono durante la gravidanza (per esempio, la rosolia).
- Madre di età superiore ai 40 anni: L'età materna avanzata può aumentare il rischio.
- Diabete mellito materno: Una condizione non controllata può influenzare lo sviluppo fetale.
- Assunzione di idantoina: Da parte di una donna in gravidanza per attenuare le crisi epilettiche; il bambino nasce con la sindrome fetale da idantoina.
- Assunzione di farmaci: Alcuni farmaci possono essere teratogeni.
Nel complesso, si ritiene che la tetralogia di Fallot possa essere il risultato di una combinazione di fattori genetici ed ambientali, in molti casi di difficile individuazione.
Grazie alle moderne tecniche diagnostiche, si è individuato che il processo che origina le quattro anomalie cardiache della tetralogia di Fallot è uno scorretto allineamento del setto aortico-polmonare durante la fase embrionale di formazione del cuore. Questo setto, che separa il cuore destro dal cuore sinistro, l'aorta dalle arterie polmonari e i due ventricoli, se malallineato, determina le prime tre anomalie sopradescritte. La quarta anomalia, l'ipertrofia del ventricolo destro, è una conseguenza delle altre tre, una risposta del cuore alla resistenza anomala.
Sintomi e Segni della Tetralogia di Fallot
I sintomi della tetralogia di Fallot possono comparire subito dopo la nascita o nei primi mesi di vita. Le anomalie anatomiche del cuore determinano la formazione e l'immissione di sangue misto (ossigenato e non ossigenato) in circolo. Il sangue misto, raggiungendo i tessuti del corpo umano, determina il principale segno della tetralogia di Fallot: la cianosi.
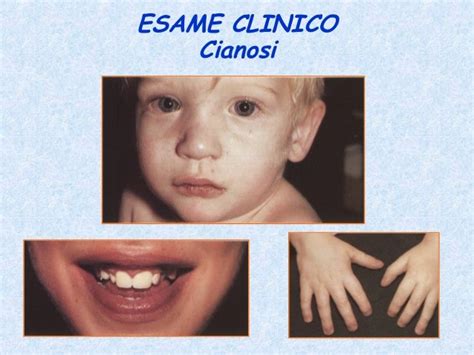
Cianosi e Crisi Ipercianotiche
La cianosi è la conseguenza clinica di un'ipossiemia cronica, ovvero la presenza in circolo di sangue poco ossigenato. Si manifesta come una colorazione bluastra della pelle e delle mucose, in particolare di labbra, estremità delle dita, dei lobi delle orecchie e della punta del naso, rendendo conto del nome popolare dato alla condizione di "sindrome del bambino blu" o "malattia del bambino blu". Maggiore è il travaso di sangue non ossigenato dal ventricolo destro verso il ventricolo sinistro ("shunt da destra a sinistra"), maggiore è il colorito cianotico.
Il sintomo potrebbe non essere presente alla nascita in tutti i casi; in una piccola parte di pazienti non è presente la cianosi (Fallot “rosa”). Tuttavia, da quel momento in poi, potrebbero svilupparsi episodi improvvisi di cianosi grave, chiamati crisi ipercianotiche o crisi asfittiche. Queste sono parossismi di iperpnea (respirazione rapida e profonda), irritabilità e pianto prolungato, aumento della cianosi e riduzione di intensità o scomparsa del soffio cardiaco. Le crisi si verificano il più delle volte nei lattanti, con un picco di incidenza nell'età da 2 a 4 mesi, e possono essere letali. Un grave episodio può portare a flaccidità, convulsioni e talvolta al decesso. In questi casi, la saturazione di ossigeno nel sangue arterioso può ridursi improvvisamente. Le crisi ipercianotiche possono essere scatenate da qualsiasi evento che diminuisce lievemente la saturazione di ossigeno (es. pianto, defecazione) oppure che diminuisce improvvisamente la resistenza vascolare sistemica (es. giocare, scalciare le gambe al risveglio) o con un esordio brusco di tachicardia o di ipovolemia. Il meccanismo di queste crisi, sebbene incerto, implica un aumento dello shunting destro-sinistro e una caduta della saturazione arteriosa di ossigeno, spesso a causa di un aumento dell'ostruzione del tratto di efflusso ventricolare destro, un aumento delle resistenze vascolari polmonari, o una diminuzione della resistenza sistemica.
Altri Sintomi e Segni Generali
- Basso peso alla nascita: O ritardo della crescita e dello sviluppo.
- Agitazione.
- Dispnea, affanno e fatica: In particolare, durante l'allattamento o dopo un pianto prolungato.
- Tendenza ad assumere una posizione accovacciata (squatting): Durante il gioco, alcuni lattanti possono accovacciarsi a intermittenza, una posizione che aumenta la resistenza vascolare sistemica e la pressione aortica, diminuisce lo shunting ventricolare destro-sinistro e così aumenta la saturazione arteriosa di ossigeno.
- Dita "a bacchetta di tamburo": Dovute alla mancanza di sangue ossigenato in circolo. L'aspetto delle unghie è anch'esso particolare e viene detto "a vetrino di orologio".
- Facies piangente: Dovuta alla mancanza (agenesia) o all'ipoplasia del muscolo depressore dell'angolo della bocca.
Sintomi e Segni Cardiaci
- Soffio cardiaco: I neonati con tetralogia di Fallot spesso presentano un soffio cardiaco. L'esame obiettivo prevede l’auscultazione toracica completa per mezzo dello stetofonendoscopio. Con tale manovra vengono valutate soprattutto le caratteristiche del soffio sistolico legato alla stenosi polmonare, in termini di durata, intensità o trasmissibilità. Il soffio nella tetralogia è dovuto alla stenosi polmonare; di solito il difetto del setto interventricolare (comunicazione interventricolare) è silenzioso perché è grande e non ha alcun gradiente di pressione. Pertanto, man mano che l'ostruzione all'efflusso polmonare diventa più grave, il soffio diventa più dolce. Minore è l'intensità sonora del soffio sistolico polmonare, maggiori sono la stenosi e la quantità di sangue travasato nel ventricolo sinistro, e più intenso è il colorito cianotico. Viceversa, più è forte il rumore del soffio, minori sono la stenosi e la quantità di sangue travasato nel ventricolo sinistro, e meno intenso è il colorito cianotico.
- Secondo tono cardiaco (S2) singolo: Questo è dovuto al fatto che la componente polmonare è marcatamente ridotta.
- Prominente impulso ventricolare destro e fremito sistolico.
- Aritmia: Battiti cardiaci irregolari.
- Scompenso cardiaco: In una piccola parte di pazienti non è presente la cianosi (Fallot “rosa”), ma segni di scompenso cardiaco come difficoltà all’alimentazione, ridotto accrescimento corporeo, aumento della frequenza respiratoria e cardiaca.
Sintomi e Segni Oftalmologici
Nei pazienti con tetralogia di Fallot, può osservarsi una congestione dei vasi sanguigni della retina.
Altri Disturbi Associati
In associazione alla tetralogia di Fallot, si possono presentare altre anomalie, cardiache e non. Una di queste è l'atresia polmonare, una condizione assai grave che consiste nella completa ostruzione del tratto che consente la fuoriuscita di sangue dal ventricolo destro, cioè l'arteria polmonare. In questi casi, l'unica via pervia è quella creata dal buco interventricolare; il travaso di sangue ("shunt da destra a sinistra") è completo e la cianosi diventa severa. Altre malformazioni associate comprendono:
- Difetto del setto atriale (con la sua comparsa, si parla di pentalogia di Fallot): 10% dei casi.
- Arco aortico destroposto (anziché a sinistra): 25% dei casi.
- Valvola polmonare bicuspide: 60% dei casi.
- Anormalità delle arterie coronarie: 10% dei casi.
- Atresia polmonare (o pseudo tronco arterioso): percentuale sconosciuta.
- Scoliosi: percentuale sconosciuta.
Diagnosi della Tetralogia di Fallot
La diagnosi della tetralogia di Fallot si basa su un insieme di valutazioni cliniche e strumentali. Il colorito cianotico alla nascita e il soffio sistolico polmonare sono già due indizi validi che fanno pensare alla tetralogia di Fallot. Molte cardiopatie congenite, anche gravi, sono ben tollerate durante il periodo fetale per la presenza di strutture che mettono in comunicazione il cuore destro con il cuore sinistro (forame ovale) e la circolazione polmonare con la circolazione sistemica (dotto di Botallo). Queste comunicazioni scompaiono fisiologicamente 12-24 ore dopo la nascita, modificando l’equilibrio emodinamico pre-esistente. La diagnosi avviene spesso alla nascita o nel periodo neonatale.
Diagnosi Prenatale
- Ecografia pre-natale e tri test: Nella maggior parte dei casi, l'ecografia morfologica permette al ginecologo di porre il sospetto di Tetralogia di Fallot. L'ecografia pre-natale e il tri test sono due esami svolti sulle madri in gravidanza, allo scopo di valutare la presenza, nel feto, di eventuali anomalie cromosomiche, come la Sindrome di Down, patologia spesso associata alla tetralogia di Fallot. I vantaggi sono che non sono nocivi né per la madre, né per il feto.
- Ecocardiografia fetale: La definitiva conferma della diagnosi si ottiene con un ecocardiogramma fetale eseguito dal cardiologo pediatra. Questo esame è d'elezione per la tetralogia di Fallot; i suoi vantaggi includono velocità, assenza di radiazioni nocive e la possibilità di svolgerlo in età pre-natale. Tramite l'ecocardiografia, fetale e trans-toracica, il medico ottiene una descrizione esauriente dell'anatomia interna del cuore: stato del setto interventricolare, stenosi della valvola polmonare, posizione dell'aorta, ipertrofia del ventricolo destro. Una buona valutazione ecografica prenatale consente di fare diagnosi prima che le fisiologiche modifiche post-natali del circolo si instaurino e rendano evidente la malformazione, così da consentire un tempestivo trattamento di cardiopatie altrimenti incompatibili con la vita e dunque una prognosi migliore. In casi selezionati è possibile una correzione chirurgica intrauterina.
Diagnosi Postnatale
Dopo la nascita, la diagnosi si avvale dell’esame clinico da parte del neonatologo prima, del pediatra poi, ed eventualmente del cardiologo esperto in patologie pediatriche.
- Pulsossimetria (screening neonatale): Di grande importanza è lo screening neonatale con pulsossimetro che, misurando la saturazione di ossigeno dell’emoglobina, evidenzia precocemente una condizione di ipossia, prima ancora che sia evidente la cianosi, segno di una cardiopatia congenita critica o complessa. La tetralogia di Fallot può essere rilevata mediante uno screening neonatale condotto attraverso la pulsossimetria.
- Esame obiettivo: L'auscultazione toracica completa per mezzo dello stetofonendoscopio per valutare le caratteristiche del soffio sistolico.
- Elettrocardiogramma (ECG): Il tracciato ECG mostra, in modo chiaro, l'ipertrofia ventricolare destra e una deviazione assiale destra, e può anche mostrare una dilatazione atriale destra. È un esame non invasivo.
- Radiografia del torace (RX torace): Un tempo era l'esame di maggior affidamento. Dalle immagini radiografiche, il cuore di un paziente con tetralogia di Fallot assume una forma simile a uno stivale ("cuore a forma di stivale"), con il tronco dell'arteria polmonare concavo e una diminuzione della trama vascolare polmonare. Lo spostamento a destra dell'arco aortico è presente nel 25% dei casi. Ha lo svantaggio dell'emissione di radiazioni ionizzanti nocive.
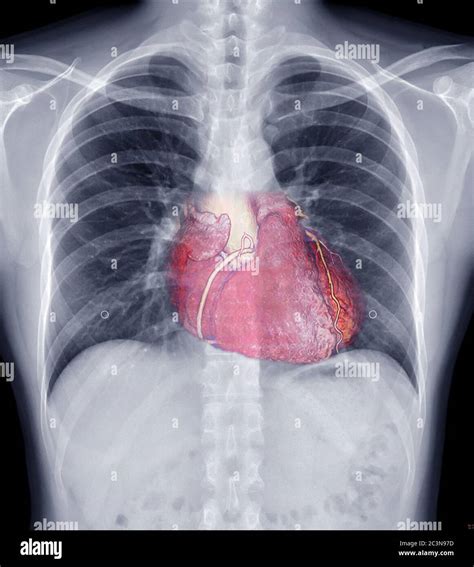
- Cateterismo cardiaco: È un esame invasivo che può essere richiesto per valutare la struttura del cuore e pianificare l’intervento chirurgico. Richiede l’inserimento di un tubo sottile e flessibile (catetere) in un vaso sanguigno, di solito introdotto dall’inguine, per poi guidarlo fino al cuore. Fornisce informazioni sull'anatomia delle cavità interne del cuore e sui livelli di ossigeno presenti nel sangue. È raramente necessario, a meno che non vi sia il sospetto di un'anomalia dell'arteria coronaria che potrebbe influenzare l'approccio chirurgico (p. es., la discendente anteriore derivante dalla coronaria destra) che non può essere chiarita con l'ecocardiografia. I cateteri possono ledere la parete dei vasi sanguigni.
- Risonanza Magnetica (RM) o Tomografia Computerizzata (TC): Possono anche essere utilizzate per evidenziare l'anatomia delle coronarie e per misurazioni accurate della funzione e volumetria del VD.
Trattamento della Tetralogia di Fallot
Il trattamento della tetralogia di Fallot è chirurgico e rappresenta l’unica possibilità di correzione definitiva. L’operazione viene generalmente eseguita tra i 3 e i 6 mesi di vita, ma può essere anticipata nei casi più gravi. Tutti i bambini con tetralogia di Fallot hanno bisogno di almeno un intervento chirurgico.
Terapia d'Emergenza per Crisi Ipercianotiche
Neonati e bambini con cianosi severa e con crisi asfittiche richiedono alcune cure d'emergenza. Sono interventi da praticare in modo immediato, poiché la vita dei pazienti è a forte rischio. In simili circostanze, le misure terapeutiche consistono nell'ossigeno-terapia e nella somministrazione di determinati farmaci.
- Posizionamento: Sistemare i lattanti in una posizione genupettorale (i bambini più grandi di solito si accovacciano spontaneamente e non sviluppano crisi ipercianotiche).
- Tranquillità: Stabilire un ambiente tranquillo.
- Ossigeno: Somministrare ossigeno supplementare.
- Liquidi EV: Somministrare liquidi EV per l'espansione del volume.
- Farmaci: La terapia medica standard comprende morfina, fenilefrina e beta-bloccanti (propranololo o esmololo). Casi clinici descrivono l'uso di midazolam e fentanil intranasali come alternative alla morfina per la gestione delle crisi asfittiche ipercianotiche, con il vantaggio di non richiedere l'accesso EV. Il bicarbonato di sodio, 1 mEq/kg EV, può essere somministrato se è presente l'acidosi metabolica.
- Prostaglandina E1: Per i neonati sintomatici con grave cianosi, la prostaglandina E1 in infusione (a partire da 0,05-0,1 mcg/kg/min EV) viene utilizzata per riaprire il dotto arterioso e quindi aumentare il flusso sanguigno polmonare.
- Misure estreme: Se il posizionamento e i farmaci non alleviano la crisi o se il neonato sta rapidamente peggiorando, possono essere necessarie l'intubazione tracheale con paralisi muscolare e anestesia generale, l'ossigenazione extracorporea a membrana, e un intervento chirurgico d'urgenza. Il propranololo da 0,25 a 1 mg/kg per via orale ogni 6 h può prevenire le recidive; la maggior parte degli esperti ritiene che anche una sola crisi significativa denoti la necessità di una rapida riparazione chirurgica.
Procedure Palliative
In alcuni neonati con basso peso alla nascita o complessa anatomia, una palliazione iniziale può essere preferita alla riparazione completa. L'obiettivo della palliazione è aumentare il flusso polmonare anterogrado per migliorare l’ossigenazione sistemica e favorire la crescita delle arterie polmonari. Si tratta di un'operazione dagli effetti temporanei, in attesa che il cuore del bambino si accresca e sia pronto per la riparazione intracardiaca.
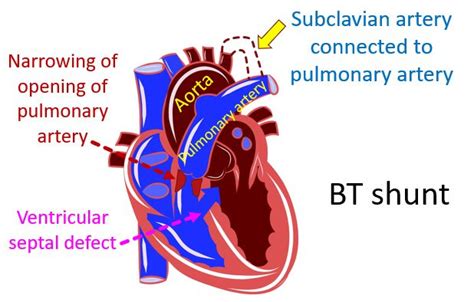
- Shunt di Blalock-Taussig modificato: La procedura abituale è uno shunt modificato di Blalock-Taussig-Thomas, nel quale l'arteria succlavia è connessa all'arteria polmonare ipsilaterale con una protesi sintetica. Questo intervento non richiede di essere condotto a cuore aperto e prevede il posizionamento di uno piccolo tubicino di materiale sintetico tra un’arteria corporea (o l’aorta) e l’arteria polmonare. Lo shunt viene chiuso al momento della riparazione completa.
- Angioplastica transcatetere o stent nel dotto arterioso: Più recentemente, l'angioplastica transcatetere del tratto di efflusso ventricolare destro o del dotto arterioso pervio è stata usata come procedura palliativa alternativa con buoni risultati. Un stent di metallo può essere posizionato nel dotto arterioso di Botallo (un vaso sanguigno tipico del feto che collega l’aorta all’arteria polmonare e che normalmente andrebbe incontro a chiusura spontanea durante il primo giorno di vita) per fornire un flusso sanguigno extra ai polmoni.
Riparazione Intracardiaca Definitiva
La riparazione intracardiaca è l'unica terapia in grado di risolvere le malformazioni cardiache della tetralogia di Fallot. L'operazione si svolge a cuore aperto. In genere, la correzione chirurgica viene eseguita intorno ai sei mesi di vita, mentre nelle forme più gravi può essere necessario un intervento precoce. Si consiglia di praticarla su pazienti di almeno un anno di età, ma alcuni casi gravi impongono un'operazione anche al sesto o nono mese di vita. La correzione chirurgica della tetralogia di Fallot viene solitamente eseguita durante il primo anno di vita.
La riparazione completa della tetralogia di Fallot consiste in:
- Chiusura del difetto del setto interventricolare: Con patch (cerotto) protesico o in pericardio autologo.
- Allargamento del tratto di deflusso ventricolare destro: Con resezione muscolare e valvuloplastica della polmonare, e, se necessario, un patch limitato attraverso l'anulus polmonare oppure sull'arteria polmonare principale. Se vi è una significativa ipoplasia dell'anulus della valvola polmonare, viene applicato un cerotto transannulare. L’intervento di correzione, infatti, avendo come obiettivo quello di ampliare il tratto di efflusso del ventricolo destro, può rendere, nel tempo, la valvola polmonare incontinente: il sangue riesce ad essere pompato dal ventricolo destro nei polmoni, ma una parte torna indietro, rigurgita, nel ventricolo destro.
L'intervento chirurgico in genere si effettua elettivamente all'età di 2-6 mesi, ma se i sintomi sono presenti o l'ostruzione nel tratto di efflusso del ventricolo destro è grave, può essere eseguito in qualsiasi momento. L'approccio transventricolare classico prevede l'incisione longitudinale del corpo del VD per la chiusura del DIV. L'approccio transatriale, un'alternativa, si effettua con incisione della parete atriale destra, preservando l'integrità anatomica del VD. L’orientamento attuale è quello di preservare per quanto possibile la continenza della valvola polmonare, con tecniche di dilatazione e ricostruzione.
Se la riparazione intracardiaca ha successo, aumentano i livelli di sangue ossigenato circolante e si riducono la cianosi e le crisi asfittiche.
Prognosi e Follow-up a Lungo Termine
La prognosi della tetralogia di Fallot è molto migliorata negli ultimi decenni grazie ai progressi della cardiochirurgia pediatrica. La mortalità perioperatoria della tetralogia di Fallot non complicata con riparazione completa è inferiore al 5%. Per i pazienti non trattati, i tassi di sopravvivenza sono del 75% al primo anno di vita, del 60% a 4 anni e del 30% a 10 anni, e solo il 5% sopravvive oltre i 40 anni. A queste percentuali, va aggiunto che un bambino non trattato soffre di uno sviluppo ritardato e che ogni sua attività fisica (anche moderata) crea affanno.
La percentuale di sopravvivenza dopo correzione chirurgica di TF è attualmente superiore al 95-97%, con risultati di sopravvivenza a distanza di 20 anni superiori al 90%. Al giorno d'oggi, più del 90% dei sopravvissuti all'intervento cardiochirurgico sono vivi a 25 anni dall'intervento. La maggior parte dei bambini operati può condurre una vita attiva e pressoché normale, sebbene con la necessità di periodiche visite di controllo per monitorarne i progressi ed eventuali future necessità.
Nonostante l'eccellenza dei risultati chirurgici, la chirurgia non "cura" la malattia nel senso di renderla completamente normale, e il cuore dei pazienti con TF operata rimane anatomicamente, fisiologicamente ed elettricamente anormale. La sopravvivenza a distanza di tali pazienti differisce da quella della popolazione generale. Si stima che per un adulto di circa 30 anni con TF operata vi sia un rischio di morte annuo pari allo 0.5% (3 volte superiore rispetto alla norma per un maschio, 8 volte superiore se femmina). Inoltre, tale rischio aumenta con l’avanzare dell’età, con un rischio annuale di mortalità che aumenta dello 0.1% ogni decade.
Le complicazioni a lungo termine più frequenti nel paziente con TF operata includono:
- Aritmie sopraventricolari e ventricolari: Sono la problematica più seria e un rischio di morte improvvisa. Circa il 34% degli adulti con TF operata presenta insorgenza di tachiaritmie atriali o sopraventricolari sintomatiche, mentre circa l’8.5% sviluppa tachicardia ventricolare sostenuta. Un crescente numero di pazienti richiede l’impianto di defibrillatore, a causa del rischio stimato di morte improvvisa pari al 2% per decade. Il QRS >180 ms è stato identificato come un significativo indice predittivo di morte improvvisa aritmica.
- Insufficienza polmonare (IP) cronica: Che conduce a dilatazione ventricolare destra e insufficienza tricuspidale. È la più frequente anomalia fisiopatologica residua.
- Anomalie della cinetica della parete ventricolare destra: Con progressiva disfunzione del VD.
- Ostruzione residua o aneurismi dell’efflusso ventricolare destro, o DIV residuo.
- Reinterventi: Durante l’infanzia, circa il 5% richiede un reintervento chirurgico e un ulteriore 6% viene sottoposto ad una procedura interventistica. Il follow-up a lungo termine indica un rischio annuo pari allo 0.8% di necessità di sostituzione valvolare polmonare (SVP), con una maggiore incidenza nei pazienti con TF e atresia polmonare o aplasia polmonare.
I pazienti operati potrebbero dover limitare l’attività fisica, in particolare per gli sport agonistici, qualora dovesse essere presente un’ostruzione residua o una perdita nella valvola polmonare, evento comune anche dopo la riparazione. In alcuni casi di ridotta funzionalità cardiaca o disturbi del ritmo potrebbero essere invitati a limiti ulteriori, ma in molti casi è possibile permettere una piena partecipazione alle normali attività di gioco e sportive.
La ricerca è focalizzata sugli esiti a distanza e sulla qualità di vita di questi pazienti. Molti studi clinici hanno indicato come la severa dilatazione e disfunzione del VD siano i più importanti fattori di rischio per eventi avversi a distanza. L'ipertrofia del VD dopo correzione della TF è in realtà dovuta non solo all’incremento del volume dei miociti, ma anche all’aumento della fibrosi e della concentrazione di collagene, quale risultato di un processo di maladattamento in risposta a un prolungato sovraccarico di volume o pressione. Le moderne tecniche chirurgiche di preservazione della valvola polmonare al momento della correzione primaria potrebbero modificare positivamente la prognosi a distanza dei pazienti con tetralogia di Fallot dopo correzione. La profilassi dell'endocardite è raccomandata prima dell'intervento chirurgico ma è necessaria solo per i primi 6 mesi dopo la riparazione, a meno che vi sia un difetto residuo adiacente a un patch chirurgico o materiale protesico.