Le aneuploidie cromosomiche rappresentano un vasto gruppo di anomalie genetiche che influenzano in modo significativo la salute e lo sviluppo degli individui. Sono condizioni genetiche caratterizzate da un numero anomalo di cromosomi in una cellula, ovvero un numero diverso da 46, che è il corredo cromosomico standard per l'essere umano. Questa condizione può avere implicazioni significative per la salute, lo sviluppo e il benessere generale di un individuo. Comprendere l'aneuploidia è fondamentale non solo per le persone colpite, ma anche per gli operatori sanitari, poiché può influenzare le decisioni terapeutiche e le strategie di gestione.
L'aneuploidia si riferisce a una condizione in cui il numero di cromosomi in una cellula non è un multiplo esatto del numero aploide, che negli esseri umani è 23. Invece dei normali 46 cromosomi (23 coppie), un individuo con aneuploidia può avere uno o più cromosomi in più (trisomia) o uno o più cromosomi mancanti (monosomia). Le aneuploidie sono anomalie del numero dei cromosomi autosomici e sessuali e, in molti casi, possono derivare da errori durante la divisione cellulare, in particolare durante la meiosi, il processo che produce spermatozoi e ovociti.
Comprendere le Aneuploidie Cromosomiche: Definizione e Classificazione
Le aneuploidie sono anomalie cromosomiche caratterizzate da alterazioni del numero dei cromosomi, cioè da un numero maggiore o minore di cromosomi rispetto al numero standard. Sono comprese le sindromi caratterizzate da anomalie del numero dei cromosomi, in cui il numero dei cromosomi è diverso da 46. Le aneuploidie dei cromosomi autosomici sono tra le anomalie cromosomiche clinicamente più importanti. Esse sono essenzialmente monosomie, ovvero la presenza di una sola copia di un cromosoma in una cellula per il resto diploide, e trisomie, che indicano la presenza di tre copie di un cromosoma.
Il termine “trisomia” identifica la presenza di tre, anziché di due, copie di un determinato cromosoma. Il termine “monosomia”, invece, identifica l’assenza di una delle due copie di un cromosoma. Le anomalie numeriche dei cromosomi possono derivare tanto da alterazioni della meiosi durante la gametogenesi parentale, quanto da alterazioni mitotiche durante le prime fasi dello sviluppo dello zigote. Le più comuni forme di aneuploidia (trisomie e monosomie) derivano in genere da un processo di non-disgiunzione meiotica. Una più rara possibilità è quella della segregazione, per la quale uno dei due cromosomi figlio viene ritardato in anafase, entrando a far parte della cellula nella quale è migrato l'omologo.
Ogni cellula somatica del nostro corpo, cioè tutte fatta eccezione per le cellule germinali (ovociti e spermatozoi), contiene 23 coppie di cromosomi, ovvero 46 strutture presenti nel nucleo delle cellule. Durante lo sviluppo degli ovociti e degli spermatozoi possono verificarsi degli errori. Capita che vengano prodotti gameti con un numero di cromosomi diverso da 23 o con cromosomi di struttura alterata. Per esempio, un ovocita può contenere 24 cromosomi invece di 23. Se viene fecondato da uno spermatozoo che ne contiene 23, il prodotto è un embrione che contiene un cromosoma in più, cioè 47. Questo eccesso o difetto di materiale genetico comporta uno sbilanciamento grossolano del genoma, spesso incompatibile con la sopravvivenza dell’embrione, per cui la gravidanza è a rischio di aborto spontaneo.
Le anomalie di numero dei cromosomi includono anche la poliploidia, come la triploidia (69 cromosomi) o la tetraploidia (92 cromosomi). L'aneuploidia può essere omogenea, cioè tutte le cellule dell’organismo ne sono affette, oppure l’anomalia può interessare solo una parte delle cellule, una condizione nota come mosaicismo. Le aneuploidie cromosomiche possono essere omogenee, cioè tutte le cellule dell’organismo ne sono affette, oppure l’anomalia può interessare solo una parte delle cellule.
Le Poliploidie: Forme Estremamente Gravi di Aneuploidia con Scarsa Correlazione con l'Età Materna
Tra le forme più estreme di anomalie del numero dei cromosomi rientrano le poliploidie, che consistono nella presenza di corredi cromosomici completi in eccesso. Queste condizioni sono quasi sempre letali e non mostrano una correlazione diretta con l'età materna avanzata, bensì con errori precoci nella fecondazione o nella divisione cellulare embrionale.
Un corredo cromosomico triploide, di 69 cromosomi, è tra le più frequenti cause di aborto spontaneo, rappresentando il 15% di tutte le anomalie cromosomiche riscontrate nei materiali abortivi. Eccezionalmente si osserva anche tra i neonati vivi, ma in questi casi ha costantemente letalità precoce. Il rischio di ricorrenza per le gravidanze successive della stessa coppia è trascurabile, suggerendo che questi eventi siano per lo più sporadici e non legati a fattori genetici ereditari o all'età materna.
Analogamente, un corredo cromosomico tetraploide, di 92 cromosomi, è sempre incompatibile con la vita. Non si riscontra mai infatti tra i nati vivi, ma solo negli aborti spontanei, dove rappresenta il 6% delle cause cromosomiche che li determinano. Anche in questo caso il rischio di ricorrenza familiare è trascurabile, a sottolineare la natura accidentale di queste anomalie.
Le Monosomie: Quando un Cromosoma in Meno Determina Quadri Clinici Specifici
Le monosomie rappresentano la mancanza di una copia completa di un cromosoma. Per quanto riguarda gli autosomi, cioè i cromosomi non sessuali, le monosomie non in mosaico non si osservano mai tra i nati vivi, essendo tutte letali in utero. Questa forte selezione naturale elimina precocemente la maggior parte degli embrioni con tali gravi anomalie.
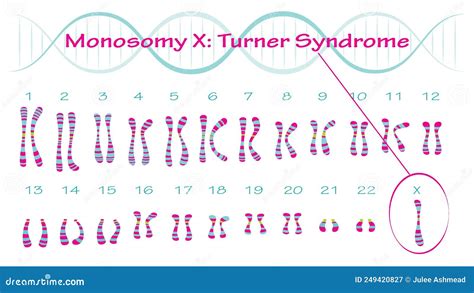
L’unica eccezione a questa regola è rappresentata dalla monosomia X, ovvero il cariotipo 45,X, che è la causa della Sindrome di Turner. Anche la monosomia X subisce tuttavia una forte selezione negativa in utero; infatti il 99% degli zigoti con cariotipo 45,X viene abortito spontaneamente entro il primo trimestre di gravidanza. Tutti i casi di monosomia X sono sporadici nelle famiglie per cui non c’è rischio di ricorrenza in gravidanze successive della coppia. Questo indica che la monosomia X non è primariamente correlata con l'età materna avanzata come fattore di rischio di ricorrenza.
Le Trisomie Autosomiche Compatibili con la Vita e le Loro Variabili Correlazioni con l'Età Materna
Le uniche trisomie, non in mosaico, degli autosomi osservabili in neonati vivi sono la trisomia 21 (sindrome di Down), la trisomia 18 (sindrome di Edwards) e la trisomia 13 (sindrome di Patau). Queste sono le aneuploidie autosomiche più studiate e clinicamente rilevanti. È in questo contesto che emerge la complessità della correlazione con l'età materna, con alcune trisomie che mostrano un chiaro legame e altre che ne sono indipendenti.
Trisomia 21: La Sindrome di Down e il Suo Forte Legame con l'Età Materna
La trisomia 21, conosciuta anche come Sindrome di Down, è causata dalla presenza di una copia in più del cromosoma 21. Rappresenta la causa genetica più comune di ritardo mentale ed anche il disordine cromosomico più frequente nell’uomo. È una delle aneuploidie più frequenti con un’incidenza, a livello mondiale, di circa 1 su 700 tra i nati vivi.
I segni clinici più significativi della sindrome di Down includono ipotonia, presente fin dalla nascita, un aspetto peculiare del capo e del volto con viso rotondeggiante, obliquità verso l'alto delle rime palpebrali, epicanto, naso piccolo con ipoplasia del dorso e narici anteverse, lingua grossa e tendente alla protrusione, padiglioni auricolari piccoli e accartocciati, e ipoplasia del segmento medio del viso. Circa il 50% delle persone Down ha una cardiopatia congenita, soprattutto in forma di canale atrio-ventricolare comune. In associazione possono trovarsi anche malformazioni extra cardiache, soprattutto nel tratto gastrointestinale, come atresia duodenale, ano imperforato e malattia di Hirschsprung. Inoltre, possono completare il quadro clinico la cataratta congenita, la bassa statura, la sindrome di West, le crisi epilettiche, la leucemia, le patologie autoimmuni e endocrine (ipotiroidismo, intolleranza al glutine, diabete, alopecia). Il ritardo mentale è costante, ma di grado variabile anche in assenza di mosaicismo. L'aspettativa di vita è al momento superiore ai 50 anni.
È importante sottolineare che l’età materna è un fattore determinante la probabilità di nascita di un figlio Down: si passa da una probabilità di 1/2.500 prima dei 20 anni, a una di 1/700 tra i 30 e i 34 anni, 1/230 tra i 35 e i 39 anni, 1/60 tra i 40 e i 44 anni, fino a una probabilità di circa 1/50 se l’età materna supera i 45 anni. "Questo tipo di alterazioni nella maggior parte dei casi origina nei gameti femminili", spiega il genetista Bruno Dallapiccola, direttore scientifico dell’Ospedale Pediatrico Bambino Gesù di Roma, "e il rischio per una coppia di concepire un embrione portatore di aneuploidia cresce proporzionalmente con l’aumentare dell’età materna." Non sappiamo con precisione perché ciò avvenga, ma la ragione è probabilmente legata alle caratteristiche dell’oogenesi, cioè della formazione degli ovociti, un processo che può durare decenni, dato che inizia nella vita embrionale, per poi bloccarsi e sbloccarsi ciclicamente solo dopo la pubertà. Per questo il concepimento in età femminile avanzata riguarda una cellula che ha iniziato la sua prima divisione decenni prima e che è stata esposta per anni a potenziali agenti ambientali nocivi.

L'origine della sindrome di Down: cosa provoca la trisomia 21
Trisomia 18: La Sindrome di Edwards e la Sua Correlazione con l'Età Materna
La trisomia 18, conosciuta anche come Sindrome di Edwards, viene causata da una copia in più del cromosoma 18. Presenta una frequenza di 1/6000-1/8000 nati, è più frequente nelle femmine ed è correlata con età materna avanzata. Ha un alto rischio di abortività.
I segni clinici specifici includono un marcato ritardo di crescita prenatale e postnatale, microcefalia, un aspetto peculiare del volto, che è minuto con naso sottile, microretrognazia, orecchie a basso impianto e con padiglioni a punta. Molto frequenti sono le malformazioni congenite, soprattutto cardiovascolari (in oltre il 90% dei casi), ma anche scheletriche, gastrointestinali e renali. Nelle prime settimane di vita sono presenti ipotonia, iporeattività e difficoltà alimentari (scarsa suzione), seguite dalla progressione verso l’ipertono con l’apparente perdita della percezione dell’ambiente circostante. Il ritardo psicomotorio è molto grave, e la mortalità è del 90% entro il primo anno di vita.
Trisomia 13: La Sindrome di Patau e la Sua Indipendenza dall'Età Materna
La trisomia 13, conosciuta anche come Sindrome di Patau, è caratterizzata dalla presenza di una copia in più del cromosoma 13. La T13 ha una frequenza di 1/8000-1/15000 nati, e, a differenza della trisomia 21 e 18, non è influenzata dall’età materna. Presenta, come nella trisomia 18, un alto tasso di abortività.
I segni clinici distintivi includono labiopalatoschisi, oloprosencefalia (assente o parziale separazione degli emisferi cerebrali), polidattilia e macroftalmia, oltre che anomalie dei padiglioni auricolari, anomalie scheletriche e convulsioni. Frequenti sono le malformazioni degli organi interni, a carico di cuore, rene e intestino. Il ritardo psicomotorio è molto grave, e la sopravvivenza in genere è limitata al primo anno di vita. La trisomia 13 libera riguarda circa il 75% dei casi. Nel 20% dei casi, la trisomia 13 si associa alla traslocazione Robertsoniana nella quale il cromosoma soprannumerario 13 è attaccato a un altro cromosoma acrocentrico (cromosoma 13, 14, 15, 21 o 22). Nei feti affetti la morte endouterina si verifica in oltre il 95% dei casi.
Altre Trisomie Autosomiche in Mosaicismo
Le trisomie di altri autosomi, oltre a quelle sopradescritte (21, 18, 13), sono osservabili tra i nati vivi solo in forma di mosaicismo con cellule normali. I mosaicisti cromosomici presentano in genere alcune peculiarità, quali la diversa distribuzione tissutale della popolazione cellulare trisomica, a volte assente nel sangue, e l’ampia variabilità fenotipica, che dipende in parte dalla percentuale di cellule trisomiche nell’organismo (Ad Es: Trisomia 8 costituzionale in mosaico, Sindrome di Warkany).
Le Aneuploidie dei Cromosomi Sessuali: Impatto sulla Fertilità e Quadri Clinici Sfumati
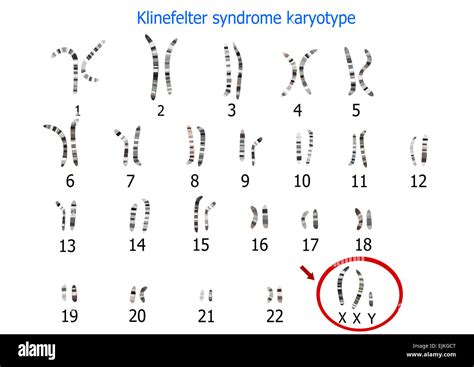
Le anomalie del numero di cromosomi sessuali in genere non si associano a segni e sintomi tipici di una sindrome cromosomica, cioè a ritardo mentale e anomalie fenotipiche multiple, quanto a disturbi della fertilità. Tra queste ritroviamo la Sindrome di Klinefelter (47, XXY), le Femmine triplo X (47, XXX), la Sindrome di Turner (45,X) e i Maschi (47, XYY).
Le aneuploidie dei cromosomi sessuali comportano quadri clinici sfumati che in molti casi si sovrappongono alla normalità. È il caso della femmina triplo-X e del maschio YY, mentre il cariotipo XXY si associa a ipogonadismo nel contesto della sindrome di Klinefelter. Queste condizioni, pur avendo implicazioni cliniche, sono spesso meno gravi in termini di ritardo mentale e malformazioni multiple rispetto alle trisomie autosomiche più comuni e la loro incidenza non è sempre strettamente legata all'età materna avanzata come per la Sindrome di Down.
Origine delle Aneuploidie: Meccanismi Genetici e Fattori di Rischio Variabili
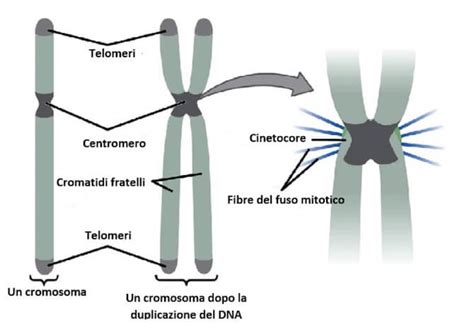
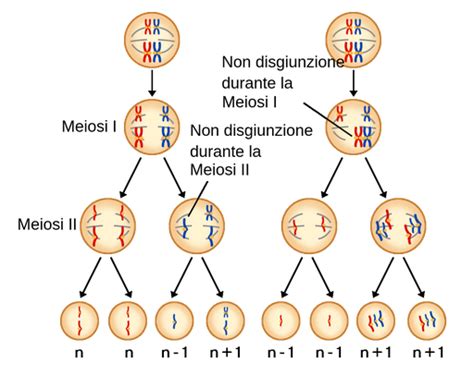
Le aneuploidie possono derivare da errori durante la divisione cellulare, in particolare durante la meiosi, il processo che produce spermatozoi e ovociti. Le più comuni forme di aneuploidia (trisomie e monosomie) derivano in genere da un processo di non-disgiunzione meiotica. Ciò avviene quando una coppia di cromosomi omologhi non si separa correttamente durante la meiosi I, o quando i cromatidi fratelli non si separano durante la meiosi II. Questo porta alla formazione di gameti con un numero anomalo di cromosomi.
Come accennato, l'età materna avanzata è uno dei fattori di rischio più significativi per l'aneuploidia, in particolare la trisomia 21 (sindrome di Down) e la trisomia 18 (sindrome di Edwards). Le ragioni di questa correlazione sono complesse e si ritiene siano legate all'oogenesi, un processo che inizia nella vita embrionale femminile e si completa solo dopo la pubertà, con gli ovociti che rimangono in uno stato di stasi per decenni, potenzialmente esposti a danni cumulativi. Diverso è il caso della spermatogenesi, che è un processo continuo nel maschio. Le divisioni cellulari che portano alla maturazione degli spermatozoi costituiscono un potenziale fattore di rischio per nuove mutazioni genetiche, ma non cromosomiche nel senso di aneuploidie numeriche.
Oltre all'età materna, altre condizioni possono influenzare il rischio di aneuploidia. Predisposizioni genetiche, come una storia familiare di anomalie cromosomiche, possono aumentare la probabilità di aneuploidia. Nella maggior parte dei casi, le aneuploidie originano da una nuova mutazione e perciò non sono presenti nel corredo cromosomico dei genitori. Può tuttavia verificarsi occasionalmente la ricorrenza dell’aneuploidia nella famiglia. Di solito ciò dipende dalla presenza di una traslocazione bilanciata in uno dei genitori, cioè una anomalia cromosomica strutturale che non comporta perdita o eccesso di materiale genetico, o più raramente dalla presenza dell’aneuploidia in mosaico in uno dei genitori.
Alcuni fattori ambientali e infezioni durante la gravidanza possono anche aumentare il rischio di anomalie cromosomiche. Ad esempio, infezioni materne come la rosolia o il citomegalovirus (CMV) possono potenzialmente portare ad aneuploidia nel feto in via di sviluppo. Infine, alcune scelte di vita e abitudini alimentari possono influenzare il rischio di aneuploidia. Ad esempio, l'obesità materna, il fumo e il consumo eccessivo di alcol durante la gravidanza sono stati associati a un aumento del rischio di anomalie cromosomiche.
Diagnosi delle Aneuploidie: Dalla Valutazione Clinica ai Test Specifici
La diagnosi di aneuploidia inizia in genere con una valutazione clinica approfondita, che include un'anamnesi dettagliata del paziente e un esame obiettivo. Quando il fenotipo è molto caratteristico, permette già alla nascita l’orientamento diagnostico. La diagnosi viene effettuata comunque in base all’esame del cariotipo, che esamina il numero e la struttura dei cromosomi in un campione di sangue o tessuto. È consigliabile l’esecuzione di una consulenza genetica da parte dei genitori del soggetto affetto.
Screening Prenatale Non Invasivo (NIPS)
Negli ultimi anni, le analisi di screening effettuate sul DNA fetale presente nel circolo materno (NIPS) hanno largamente soppiantato le tecniche invasive. Questa metodica, ultima arrivata in campo di diagnosi prenatale non invasiva, permette l'analisi diretta del DNA fetale circolante nel sangue materno. Il test ha un’attendibilità superiore al 99% ed è eseguibile sia in caso di gravidanze singole che gemellari, anche se ottenute con tecniche di procreazione assistita.
Il test Prenatalsafe® è un test di screening prenatale non invasivo disponibile in nove versioni con livelli di approfondimento diversi, che permette di rilevare nel feto aneuploidie e alterazioni cromosomiche correlate a patologie genetiche quali trisomie (come la trisomia 21 associata alla sindrome di Down), microdelezioni e alterazioni strutturali. Il DNA fetale libero circolante viene isolato dalla componente plasmatica del sangue materno e sottoposto a sequenziamento massivo parallelo (MPS) utilizzando sequenziatori Next Generation Sequencing (NGS) ILLUMINA e Thermofisher. Prenatalsafe® è sicuro per la gestante e per il feto, poiché non comporta alcun rischio di aborto come invece può accadere con le tecniche di diagnosi prenatale invasiva come l’amniocentesi o la villocentesi. Il test ha una sensibilità del 99,9% per i cromosomi 21, 18, 13 e aneuploidie dei cromosomi sessuali, con una percentuale di falsi positivi inferiore allo 0,1%. Grazie all’utilizzo di tecnologie certificate, le analisi sul DNA fetale di Eurofins Genoma hanno una sensibilità e specificità superiore al 99% su tutti i cromosomi del feto, sia per quanto riguarda l’analisi delle aneuploidie più comuni che per le anomalie cromosomiche più rare. Prenatalsafe® è inoltre rapido, con i risultati disponibili in soli 3-4 giorni lavorativi grazie alla tecnologia FAST.
Il Vera Prenatal Test® è un Test prenatale non invasivo che permette l’analisi del DNA fetale libero circolante, isolato da un campione di sangue materno, e valuta la presenza delle aneuploidie fetali comuni in gravidanza dei cromosomi 21, 18, 13 e dei cromosomi sessuali (X e Y), consentendo la determinazione del sesso fetale (opzionale). Il Vera+ Plus, in aggiunta, prevede la possibilità, ove espressamente richiesto, di eseguire un approfondimento di secondo livello, per l’individuazione nel feto delle aneuploidie a carico di ogni cromosoma e delle alterazioni cromosomiche strutturali.
Test di Screening Prenatale Meno Recenti
Nel tempo sono stati sviluppati diversi test di screening per la sindrome di Down e altre aneuploidie, sebbene con un’attendibilità inferiore rispetto all'NIPS e con una maggiore incidenza di falsi positivi o negativi.Il Tri-Test, ora considerato obsoleto, veniva effettuato tra la 15a e la 17a settimana di gestazione. Prevedeva il dosaggio di tre sostanze prodotte dal feto e dalla placenta che si ritrovano nel sangue materno: le alfa-feto-proteine (AFP), l’estriolo non coniugato (uE3) e la gonadotropina corionica umana (hCG). Dalla combinazione statistica di questi parametri ematici con l’età materna, il suo peso corporeo, l’esatta settimana di gravidanza e i dati ecografici dello sviluppo del feto, veniva formulata una percentuale di rischio. Un test positivo non indicava che il feto fosse malato, ma identificava una donna con aumentato rischio.Il Test Combinato (biochimico-ecografico) del secondo trimestre, elaborazione del Tri-Test, è finalizzato allo screening per la sindrome di Down, trisomia 18 e difetti del tubo neurale. Consiste nel prelievo di sangue materno per il dosaggio di AFP e free beta HCG e nella misura ecografica della lunghezza del femore (LF) e del diametro biparietale (BPD).Per una diagnosi precoce, in epoca compresa tra la 10a e la 13a settimana di gestazione, possono essere eseguiti il Bi-Test e la Translucenza Nucale (NT). Il Bi-Test consiste nel dosare su sangue materno due sostanze denominate free beta HCG e PAPP-A. La Nuchal Translucency (NT) è un sottile spazio fluido nella regione nucale fetale, il cui aumento è correlato ad anomalie cromosomiche. Questo screening ecografico, da effettuarsi tra l'11a e la 13a settimana, è in grado di identificare circa il 70-80% dei feti affetti. Nelle gravidanze gemellari e nelle donne diabetiche insulino-dipendenti, l’NT è il test con maggiore predittività e attendibilità. Il Test Combinato (biochimico-ecografico) del primo trimestre combina il dosaggio di PAPP-A e free beta HCG con la misura ecografica della NT, raggiungendo un’attendibilità del 90%.
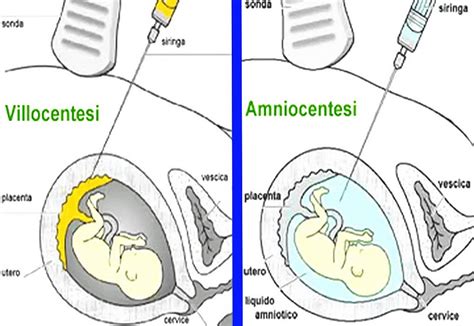
Diagnosi Prenatale Invasiva: Villocentesi e Amniocentesi
L’analisi citogenetica delle cellule fetali permette di individuare, già in epoca prenatale, le anomalie cromosomiche del nascituro. Tuttavia, queste tecniche sono gravate da un rischio tecnico di abortività. Circa il 3% dei bambini nasce con un difetto congenito, ossia con un’anomalia insorta durante la gravidanza.Il prelievo di villi coriali (villocentesi) si esegue tra la 10a e la 12a settimana di gravidanza. Sotto diretto controllo ecografico, si procede all’introduzione di un ago che, attraversata la parete addominale materna, raggiunge la placenta e ne aspira alcuni frammenti. Il rischio di aborto è stimato nell’ordine del 3-4%.L’amniocentesi viene svolta tra la 14a e la 18a settimana. Sotto guida ecografica continua, si prelevano 15-20cc di liquido amniotico. Una parte del liquido prelevato viene di norma utilizzata per la valutazione della quantità dell’alfa feto proteina (AFP), sostanza che può essere elevata in presenza di alcune anomalie fetali come la spina bifida. Il rischio legato all’amniocentesi è stimato, invece, attorno allo 0.5-1%.Anche quando l’esame dei villi coriali o l’amniocentesi esclude la presenza di alterazioni cromosomiche nel feto, è possibile che durante il resto della gravidanza o alla nascita possano essere riconosciuti nel bambino dei difetti di altra origine, non rilevabili attraverso i due esami descritti che valutano solo una parte delle cause di anomalie genetiche.
L'origine della sindrome di Down: cosa provoca la trisomia 21
Metodologie di Analisi nelle Diagnosi Invasive: Dal Cariotipo Tradizionale al Molecolare
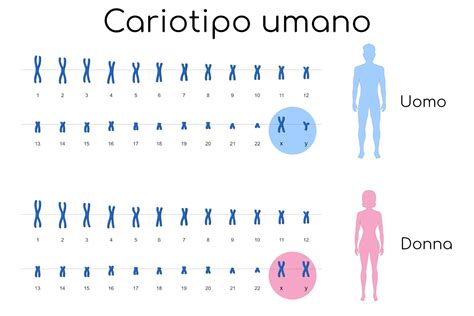
L’approccio tradizionale nella diagnosi prenatale di anomalie cromosomiche comporta la messa in coltura di cellule fetali ricavate da prelievi di liquido amniotico e la determinazione del cariotipo tramite l’analisi al microscopio dei cromosomi in metafase. Benché tale analisi sia abbastanza accurata, le colture cellulari impongono lunghi tempi di attesa che si aggirano intorno ai 15-20 giorni. Il cariotipo tradizionale, inoltre, non garantisce che il feto sia esente da malattie genetiche o alterazioni cromosomiche (delezioni o duplicazioni) di piccole dimensioni. Infatti, questo tipo di esame fornisce informazioni solo sulle principali anomalie cromosomiche (ad esempio la trisomia 21, o Sindrome di Down, le trisomie 18 e 13, la monosomia X, o Sindrome di Turner) attraverso la determinazione dell’intero assetto cromosomico fetale. Con il cariotipo tradizionale si indaga essenzialmente su quelle forme patologiche che interessano il numero e l’aspetto grossolano dei cromosomi. Nulla si potrà sapere su piccole alterazioni dei cromosomi (che sono un numero elevatissimo, anche se piuttosto rare) o sulla conformazione dei geni che sono contenuti all’interno dei cromosomi. Con il cariotipo tradizionale, infatti, si riesce ad evidenziare solo le anomalie strutturali più grandi di 10-15 Mb. Lo studio del cariotipo fetale, a differenza dell’amniocentesi rapida con la tecnica QF-PCR, presenta un’importanza diagnostica elevatissima perché evidenzia le anomalie cromosomiche più severe e frequenti (come ad esempio le trisomie) a carico di tutti i cromosomi; tuttavia, a causa dei limiti di risoluzione della tecnica, piccoli riarrangiamenti cromosomici potrebbero non essere facilmente evidenziabili.
Grazie ai recenti progressi della citogenetica molecolare è adesso possibile esaminare i cromosomi in maniera più approfondita ed accurata, utilizzando il cosiddetto Cariotipo Molecolare, procedura diagnostica che impiega una tecnica molecolare innovativa conosciuta come array-CGH. Essendo una tecnica molecolare, che non necessita di coltura cellulare, con il Cariotipo Molecolare è possibile ottenere un’analisi cromosomica approfondita in pochi giorni (3-5gg lavorativi), a differenza dei 15-20 giorni necessari con la tecnica tradizionale, riducendo al minimo i tempi di attesa dei risultati. Rispetto all’esame citogenetico tradizionale, l’analisi molecolare dei cromosomi ha una risoluzione molto più elevata (~100 volte). Ciò consente di identificare alcune patologie derivanti da alterazioni cromosomiche submicroscopiche (microdelezioni e le microduplicazioni), non evidenziabili tramite il cariotipo tradizionale, aumentando sensibilmente l’accuratezza dell’esame. Il cariotipo molecolare, infatti, consente di effettuare rapidamente non solo lo studio dell’assetto cromosomico fetale, ma anche di un gruppo di 100 patologie causate da microdelezione/microduplicazione cromosomica (es. Sindrome di Di George, la Sindrome di Williams, la Sindrome di Praeder-Willi/Angelman) ed oltre 150 geni. Inoltre, grazie ad una sofisticata analisi bioinformatica, si ha la possibilità di definire con esattezza non solo la regione genomica alterata ma anche i geni in essa contenuta, permettendo così di verificare la patogenicità dell’anomalia cromosomica riscontrata e valutare le conseguenze cliniche. L’Array-CGH è una metodica molecolare che non necessita di coltura cellulare, quindi non è soggetta al rischio di mancata crescita e, di conseguenza, di ripetizione del prelievo, garantendo un risultato nel 100% dei casi. I limiti di tale tecnica in ambito prenatale sono rappresentati dall’impossibilità di identificare riarrangiamenti cromosomici bilanciati (non patologici) e i mosaicismi (cioè la presenza di due linee cellulari con differente assetto cromosomico) con una linea cellulare scarsamente rappresentata (inferiore al 10% circa). Questa tecnica innovativa si differenzia dal cariotipo tradizionale prenatale in quanto meno laboriosa e facilmente automatizzabile, e quindi meno soggetta a rischio di errore. Inoltre, alcune sue particolarità tecniche consentono l’accertamento anche dei mosaicismi (non inferiori al 10%), e coadiuvata dalla QF-PCR permette di determinare lo stato di zigosità in gravidanze gemellari come pure la rapida identificazione di contaminazione materna che non è apprezzata dalla FISH e dal cariotipo. Il cariotipo molecolare, a differenza dell’altra tecnica di amniocentesi rapida, la QF-PCR, fornisce in tempi similmente rapidi i risultati di eventuali anomalie a carico di tutti i cromosomi.
La QF-PCR su liquido amniotico è un esame che permette di riconoscere trisomie o monosomie esclusivamente relative ai cromosomi: 13, 18, 21, X e Y, fornendo un esito preliminare in tempi rapidi (3-5gg lavorativi). Tuttavia, non è sostitutivo dell’analisi cromosomica convenzionale, non identifica con certezza trisomie o monosomie parziali dei cromosomi analizzati e può non riconoscere la presenza di mosaicismi cromosomici.
In caso di necessità diagnostica, l’amniocentesi molecolare può essere integrata dalla diagnostica prenatale Molecolare infettivologica, che consiste nell’effettuare la ricerca con tecniche molecolari della presenza del genoma di agenti infettivi, (es. Citomegalovirus, Herpes simplex, Varicella Zooster, Rubeovirus, HIV, Toxoplasma gondii, Parvovirus). Il vantaggio del ricorso alla tecnica molecolare (Polimerase Chain Reaction - PCR) risiede nel fatto che si ricerca direttamente il genoma, ossia la forma replicativa, dell’agente infettivo, superando i metodi tradizionali indiretti che esprimevano la produzione anticorpale fetale (IgM). Il tempo necessario per la diagnosi è di circa 2-3 giorni.
Le Sindromi da Microdelezione: Quando Piccole Alterazioni Hanno Grandi Effetti
Le sindromi da microdelezione sono anomalie caratterizzate dall’assenza di un tratto cromosomico di piccole dimensioni con conseguente perdita di informazione genica. Queste alterazioni causano sindromi di importanza clinica variabile a seconda del cromosoma coinvolto, della regione cromosomica interessata e delle relative dimensioni. Il Pannello Microdelezioni, aggiunto al Vera Prenatal Test® o al Vera+Plus®, valuta la presenza di alcune tra le principali sindromi da microdelezione, con una risoluzione ≥ 3,5 Mb.
Sindrome di DiGeorge (delezione 22q11.2)
È una delle più comuni sindromi da delezione cromosomica con un’incidenza mondiale di 1/2.000-1/4.000 nati vivi. Si manifesta con un quadro fenotipico estremamente variabile. Le più frequenti manifestazioni riguardano il sistema immunitario, con deficit immunitario da aplasia/ipoplasia del timo, che li rende suscettibili alle infezioni e le cardiopatie (77% dei casi) che riguardano soprattutto la regione troncoconale. Oltre il 75% dei pazienti presenta anomalie del palato. È frequente il ritardo dello sviluppo.
Sindrome di Cri-du-Chat (delezione 5p15.3)
È un disturbo genetico raro (incidenza di 1/50000) causato da una delezione del cromosoma 5 (5p-). Le caratteristiche cliniche includono dismorfismi facciali, malformazioni viscerali, microcefalia, pianto dalla tonalità acuta e monotona simile al miagolio del gatto.
Sindrome di Prader-Willi/Angelman (delezione 15q11.2-q13)
È eterogenea dal punto di vista clinico e genetico, colpendo 1/25.000 nati. La Sindrome di Prader-Willi è caratterizzata da anomalie ipotalamico-pituitarie associate a grave ipotonia nel periodo neonatale, seguita da iperfagia e mancanza di sazietà che causa obesità grave. La Sindrome di Angelman è una malattia neurologica caratterizzata da grave ritardo mentale e dismorfismi facciali caratteristici, con prevalenza tra 1/10.000 e 1/20.000. I sintomi caratteristici includono assenza del linguaggio, crisi di riso associate a movimenti stereotipati delle mani, microcefalia e disturbi neurologici con andatura da “burattino”.
Sindrome da delezione 1p36
È considerata una delle più comuni sindromi da delezione cromosomica, con un’incidenza di 1/5.000-10.000 nati vivi. I pazienti presentano caratteristici dismorfismi craniofacciali, ipotonia congenita e ritardo dello sviluppo motorio e del linguaggio.
Sindrome di Wolf-Hirschhorn (delezione 4p)
La prevalenza è di 1:50.000 nati. Si osservano marcato ritardo della crescita prenatale, una facies tipica ad “elmo da guerriero greco”, microcefalia, anomalie scheletriche e ipotonia. Il ritardo dello sviluppo è grave, con deficit cognitivo moderato-grave.
Sindrome di Jacobsen (delezione 11q23)
La prevalenza è stimata in circa 1/100.000 nati. I segni clinici più comuni sono il ritardo di crescita pre- e post-natale, il ritardo psicomotorio e i dismorfismi facciali. Circa il 20% dei bambini muore nei primi due anni di vita, spesso per le complicanze della cardiopatia.
Sindrome di Langer-Giedion (delezione 8q24.11-q24.13)
È caratterizzata da deficit cognitivo, associato a varie anomalie, compresa la cute ridondante, le esostosi cartilaginee multiple, la facies caratteristica e le epifisi falangeali “a cono”.
Sindrome di Smith Magenis (delezione 17p11.2)
La prevalenza mondiale è di 1/15.000-25.000. È causata dalla delezione del gene RAI1 nel 90% dei casi.
Gestione e Prognosi delle Aneuploidie
Il trattamento dell'aneuploidia dipende in larga misura dalla condizione specifica e dai sintomi associati. Sebbene non esista una terapia specifica per curare l'aneuploidia, le opzioni terapeutiche si concentrano sulla gestione dei sintomi e sul miglioramento della qualità della vita. Ciò include il monitoraggio e la cura di eventuali complicanze mediche, quali problemi motori, gastrointestinali, respiratori o epilettici.
Disturbi comportamentali e cognitivi possono necessitare di un approccio multidisciplinare e di un supporto pedagogico. È utile l’impostazione di un trattamento riabilitativo adeguato, che può includere fisioterapia, logopedia e terapia occupazionale, a seconda delle esigenze individuali. I pazienti devono essere sottoposti a un follow-up neurologico, neuropsichiatrico e riabilitativo costante per monitorare l'evoluzione della condizione e adattare i piani di intervento.
La prognosi per gli individui con aneuploidia varia notevolmente a seconda del tipo specifico di aneuploidia e dei problemi di salute associati. Le complicazioni a breve termine possono includere problemi di salute immediati, come difficoltà di alimentazione nei neonati o ritardi nello sviluppo nei bambini piccoli. Gli esiti a lungo termine per gli individui con aneuploidia variano notevolmente, e dipendono dalla gravità delle anomalie e dall'efficacia delle terapie di supporto.

tags: #aneuploidie #cromosomiche #non #correla #con #l