Nel panorama della medicina moderna, l'attesa di un bambino è un momento di grande gioia e speranza, accompagnato dal desiderio primario che sia "sano". Questo è il primo pensiero quando si aspetta un bebè. Fino a qualche decennio fa, un figlio custodito nel ventre materno era un vero e proprio mistero: nei secoli, indovini e stregoni si sono cimentati a decifrarlo, a partire dalle innumerevoli quanto inutili tecniche divinatorie per scoprire almeno il sesso del bambino. Oggi possiamo affermare di avere in parte svelato il mistero perché decodifichiamo le informazioni contenute nel materiale genetico e attraverso l’ecografia spiamo il piccolo in tempo reale.
Tuttavia, la decisione di avere un figlio, come qualsiasi cosa nella vita, non è comunque del tutto scevra da rischi. Indipendentemente dalla causa, ogni futuro genitore presenta un rischio di base del 3% di avere un bambino con un difetto congenito alla nascita più o meno grave. Difetti congeniti si riscontrano solo nel 5% delle gravidanze e possono essere più o meno gravi. È importante sapere che non tutti i difetti congeniti sono diagnosticabili, né con metodi ecografici né con indagini su sangue o liquido amniotico, e che non esiste un unico esame in grado di dirci se il bambino sarà sano o meno. Oggi alcuni di questi rischi e alcuni dei difetti congeniti del feto possono essere individuati e diagnosticati durante la gravidanza, attraverso la diagnosi prenatale.
Rispetto a qualche anno fa, la diagnosi prenatale sta cambiando ed è diventato complesso eseguire il counselling di informazione alle coppie nel primo trimestre di gravidanza. È aumentato il panorama degli esami di diagnosi prenatale non invasiva, grazie al ricorso sempre più frequente agli esami del DNA fetale, e sta diminuendo il ricorso agli esami invasivi.
Che Cos'è il Cariotipo e La Sua Rilevanza
L’analisi cromosomica o cariotipo è il test che consente di individuare la presenza di alcune anomalie cromosomiche, fondamentale nella diagnosi di malattie genetiche, di alcuni difetti congeniti o di alcune patologie a carico del sangue e del sistema linfatico. I cromosomi sono le strutture filiformi, contenute nel nucleo delle cellule, che custodiscono le informazioni genetiche dell’organismo. Il cariotipo è il corredo cromosomico di una persona, che deriva per il 50% dalla madre e per il 50% dal padre. La determinazione del cariotipo fetale costituisce un’indagine importante in quanto permette di evidenziare eventuali anomalie cromosomiche, sia numeriche (quali trisomie, monosomie e presenza di un marcatore), che strutturali (traslocazioni, delezioni ed inversioni). In questo contesto, l'analisi del cariotipo è lo studio del corredo cromosomico di un individuo.
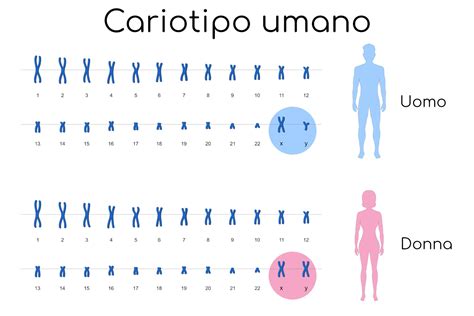
Un esame del cariotipo serve ad evidenziare la presenza di cambiamenti insoliti nei cromosomi. Nell'uomo, le aneuploidie numeriche più frequenti osservate sono la monosomia (assenza di un elemento nella coppia di cromosomi omologhi) e la trisomia (presenza di un elemento addizionale in una coppia di cromosomi omologhi). Un difetto o un eccesso dei cromosomi può provocare sintomi più o meno gravi. L’aneuploidia è causata, nella maggior parte dei casi, da errori di non-disgiunzione alla meiosi che causano la formazione di due cellule (gameti) che contengono rispettivamente un cromosoma in più ed uno in meno. La causa della non-disgiunzione non è nota, ma si verifica con maggior frequenza nella meiosi femminile ed aumenta con l’età. Da ciò deriva un aumentato rischio di patologia cromosomica fetale in madri di età superiore o uguale a 35 anni.
L'Evoluzione dei Test di Screening Prenatale
È importante sottolineare che nella diagnosi prenatale, esami e test si eseguono quando il feto è già in parte formato (la sua formazione è quasi completa, negli organi, tra la nona e la decima settimana). Pertanto, gli esami di diagnosi prenatale non aiutano a valutare le probabilità che il feto sviluppi trisomie, malattie genetiche o malformazioni, ma a diagnosticarle nel caso siano già presenti. I test di screening prenatale vengono eseguiti, generalmente, durante il primo o il secondo trimestre di gravidanza, ma non garantiscono una diagnosi certa e definitiva, ed essendo test di screening calcolano un rischio. Qualora il risultato sia a rischio (positivo), è bene rivolgersi al ginecologo o al genetista per discutere dei possibili e ulteriori esami da eseguire successivamente. Fino a poco tempo fa, i rischi connessi con l’esecuzione di esami invasivi (amniocentesi e villocentesi) orientavano i sanitari a indirizzare alla diagnosi prenatale solo la popolazione femminile selezionata come “a rischio”.
Il Test del DNA Fetale (NIPT): Uno Screening non Invasivo
Oramai abbiamo un panorama svariato di test del DNA fetale. Spesso viene chiamato con il nome delle ditte che producono il kit. La finalità del test del DNA fetale è avere una stima del rischio di aneuploidie (patologie cromosomiche). Il test del DNA fetale è un test che analizza il DNA fetale libero (cfDNA) circolante nel sangue materno per rilevare aneuploidie, alterazioni cromosomiche strutturali (delezioni e duplicazioni segmentali) fetali a carico di ogni cromosoma e gravi malattie genetiche. Lo Screening Prenatale non Invasivo su sangue materno è un’indagine eseguibile a partire dalla 10° settimana.

Il test del DNA ha una sensibilità del 99% per la trisomia 21 o sindrome di Down, del 97% per la trisomia 18 e 13. È possibile, con minor accuratezza, determinare il rischio di anomalie di numero dei cromosomi sessuali. L'accuratezza del test è molto elevata, con una precisione superiore al 99% per la rilevazione delle principali trisomie (21, 18 e 13). È un test di screening non diagnostico, effettuabile dall'11^ alla 24^ settimana. Il momento ideale per effettuare il test del DNA fetale è tra la 10ª e la 12ª settimana di gravidanza. In questo periodo, la quantità di DNA fetale libero nel sangue materno è sufficiente per garantire risultati accurati.
Sotto le spinte tecnologiche e commerciali, nel corso degli anni si è assistito all’aggiunta di altre anomalie cromosomiche, alcune sindromi da microdelezione fino al cariotipo. Attualmente, le più importanti società scientifiche riconoscono una chiara validità e utilità clinica del test del DNA limitatamente alle tre trisomie principali (T21, T18 e T13). Non tutte le gravidanze richiedono il test del DNA fetale. Il test è stato validato su gravidanze singole o gemellari, monozigotiche o dizigotiche, con circa 10 settimane di gestazione. Il test PrenatalSAFE Karyo non può escludere la presenza di tutte le anomalie cromosomiche fetali. L’esame evidenzia il 92,6% delle anomalie cromosomiche fetali che sono rilevabili in epoca prenatale ed evidenzia il 95,5% delle anomalie cromosomiche fetali rilevabili in epoca prenatale e il 99.1% di quelle riscontrate alla nascita.
Il test viene eseguito tramite un semplice prelievo di sangue materno, che viene analizzato per isolare frammenti di DNA fetale. Questi frammenti vengono poi esaminati in laboratorio per individuare eventuali anomalie cromosomiche. Il costo di Test Dna Fetale (Cariotipo Fetale) in Italia può variare a seconda della struttura sanitaria e della regione. Il test non può essere eseguito ovunque né da chiunque. La situazione italiana è molto variegata, e solo la Regione Emilia Romagna ha recentemente avviato un programma di screening che ne prevede l’introduzione in tutta la regione, dopo una fase pilota di 9 mesi limitata all’area metropolitana di Bologna. Non è una moda, perché rappresenta al momento lo standard migliore per la ricerca di alcune patologie cromosomiche importanti, e non è una necessità perché non tutte le coppie richiedono l’accesso alla diagnosi prenatale, che in prospettiva apre la strada a una possibile interruzione della gravidanza in caso di feto portatore di malattia cromosomica. Il NIPT è semplicemente un’opportunità per le coppie che desiderino effettuare l’indagine prenatale.
L'Importanza dell'Ecografia e del Bi-Test
Non c’è un solo test del DNA fetale, ma ci possono essere ampliamenti di indagine, che non si sostituiscono agli esami di screening tradizionali ma spesso hanno un significato se associati a ecografie ostetriche. È l’ecografia che permette di vedere se il feto è uno o più di uno e, in caso di malattie o malformazioni congenite in famiglia, è il momento giusto per discutere con ginecologo o genetista (se non già fatto in epoca preconcezionale) l’approccio e le opzioni di esami di diagnosi prenatale per la coppia (counseling prenatale).
Un'ecografia che si effettua tra la 11^-14^ settimana è cruciale per vedere la lunghezza del feto, lo spessore retronucale del feto (translucenza nucale), l’osso nasale, il rigurgito della tricuspide, il dotto venoso. La valutazione della translucenza nucale o NT è un dato importante per valutare il rischio fetale per le principali trisomie (13, 18, 21), ma non solo: a volte è indicativo di cardiopatie e sindromi (insieme di sintomi e segni) che si sviluppano successivamente. L’esame va eseguito in associazione a un prelievo di sangue (livelli di PAPP-A e hCG).
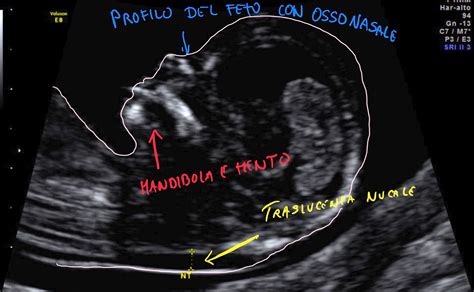
Il test del DNA fetale e il Bi Test (test combinato) sono esami prenatali che possono rilevare anomalie cromosomiche, ma differiscono per metodo, tempi e accuratezza. Il Bi Test combina un’ecografia e un’analisi del sangue per valutare il rischio di trisomia 21, 18 e 13, ma ha un’accuratezza inferiore rispetto al test del DNA fetale. Utile è associare sempre un’ecografia ostetrica in base al periodo in cui viene effettuato il test. Ad esempio, se il test del DNA fetale viene effettuato alla 12^ settimana è conveniente associare allo screening la valutazione della translucenza nucale del feto (NT), valore che rappresenta uno “spartiacque” nella diagnosi prenatale. Valori di translucenza elevata (ad esempio >3mm) richiedono esami più approfonditi.
Quando si Rende Necessaria l'Indagine Invasiva: Amniocentesi e Villocentesi
Nei casi in cui il Bitest o il test del DNA fetale libero circolante indicano un rischio aumentato per trisomia 13, 18, 21 dopo un’adeguata consulenza è possibile prendere in considerazione la possibilità di svolgere altri esami più invasivi come la villocentesi o l’amniocentesi. Questi esami presentano entrambi un lieve rischio di aborto spontaneo (circa l’1%). La Sindrome di Edwards ed altre patologie possono richiedere tale indagine. L’amniocentesi e il cariotipo vanno obbligatoriamente effettuati nelle pazienti con età maggiore ai 35 anni, in pazienti con bitest positivo, con anomalie ecografiche o con anamnesi familiare positiva per patologie ereditabili. È inoltre obbligatoria in donne di età maggiore ai33 anni con gravidanza gemellare. Data la bassa casistica di complicanze è comunque consigliata a tutte le gravide.
Amniocentesi
L’analisi del cariotipo da liquido amniotico viene effettuato mediante il prelievo di 20 ml di liquido amniotico per via trans-addominale, sotto controllo ecografico, tra la 15° e la 18° settimana di gestazione. Il liquido prelevato contiene cellule del bambino che possono essere analizzate in laboratorio.
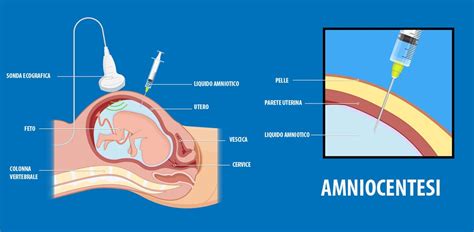
Il liquido prelevato viene centrifugato per separare la parte liquida (che verrà utilizzata per il dosaggio dell’alfafetoproteina) dalla frazione corpuscolata, costituita dalle cellule fetali che sono in sospensione nel liquido amniotico. Tali cellule, definite amniociti, sono poste in coltura con un terreno nutritivo in un adatto incubatore, alla temperatura di 37°C; in queste condizioni le cellule possono continuare a vivere al di fuori dell’organismo. Vengono allestite 3 colture distinte in contenitori sterili costituiti da una plastica particolare che lascia aderire gli amniociti e ne consente la moltiplicazione. Ciascuna cellula che aderisce alla fiasca di coltura inizia la divisione cellulare e forma un “clone” di cellule tutte uguali tra loro.
Dopo 3 giorni, la divisione cellulare viene bloccata in tutte le cellule allo stadio di metafase, con 46 cromosomi ben spiralizzati, tutti composti da due cromatidi e allineati sul piano centrale della cellula (“equatore”). A questo punto la cellula viene aperta con una sostanza ipotonica che rigonfia il nucleo e provoca la rottura della membrana nucleare. Ciò causa lo spargimento dei cromosomi nella zona del vetrino dove prima era situato il nucleo, che possono essere colorati con tecniche diverse, osservati al microscopio e fotografati. I cromosomi vengono disposti in ordine decrescente rispetto alle dimensioni, dal più grande al più piccolo, e avvicinando, a coppie, i due cromosomi uguali od omologhi. Al termine, potremo osservare che sono presenti 22 coppie di cromosomi omologhi, numerate da 1 a 22, per un totale di 44 cromosomi. I due rimanenti cromosomi, i cromosomi sessuali o “gonosomi”, sono di due tipi, detti “X” e “Y”. Il cromosoma X è piuttosto grande, di dimensioni simili a quelle del cromosoma 6, mentre il cromosoma Y è molto piccolo, solitamente di dimensioni simili ad un cromosoma 21 o 22 e contiene pochi geni.
Villocentesi
Il prelievo dei villi coriali è una procedura che prevede il prelievo di un frammento di tessuto dai villi coriali, parti della placenta materna che tuttavia di solito hanno gli stessi cromosomi del nascituro (la placenta è l’organo che cresce nell’utero materno per fornire nutrienti e ossigeno al bambino). Il cariotipo da Villi Coriali viene effettuato mediante il prelievo di 20 mg circa di villi coriali per via transaddominale sotto controllo ecografico, tra la 11° e la 13° settimana di gestazione.
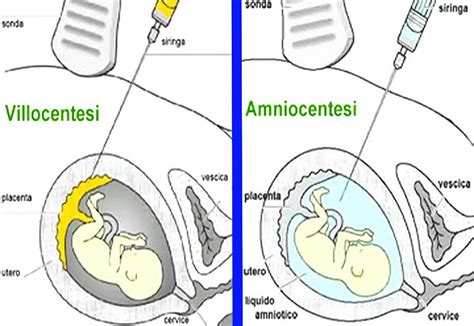
Con il metodo diretto si sfruttano le divisioni spontanee delle cellule. Un’aliquota dei villi, precedentemente puliti, verrà quindi esaminata direttamente dopo una breve incubazione a 37°C. Con il metodo della coltura, le cellule sono seminate su un apposito vetrino e incubate alla temperatura di 37°C, in modo tale da potersi moltiplicare. Vengono allestite 2 colture distinte in contenitori sterili costituiti da una plastica particolare che lascia aderire le cellule e ne consente la moltiplicazione. In questo caso ogni cellula dividendosi più volte darà origine a una colonia (un “clone” di cellule tutte uguali tra loro), cioè ad un insieme di cellule che hanno tutto lo stesso corredo cromosomico della cellula originaria. Quando i cloni sono ben sviluppati e in numero sufficiente, e ciò avviene in media in 9-15 giorni, le cellule vengono staccate e deposte su un vetrino sterile (metodica di tripsinizzazione) che favorisce ulteriormente la moltiplicazione cellulare.
In entrambe le aliquote di villi si procede con il blocco della divisione cellulare allo stadio di metafase, con 46 cromosomi ben spiralizzati, tutti composti da due cromatidi e allineati sul piano centrale della cellula (“equatore”). A questo punto le cellule vengono aperte con una sostanza ipotonica che rigonfia il nucleo e provoca la rottura della membrana nucleare. Dalla foto di un gruppo di cromosomi appartenenti ad una stessa cellula vengono ritagliate le immagini di ciascun cromosoma e in questo modo è possibile disporre l’immagine di ogni cromosoma ordinatamente su un foglio di carta, eseguendo una sorta di collage. I cromosomi vengono disposti in ordine decrescente rispetto alle dimensioni, dal più grande al più piccolo, e avvicinando, a coppie, i due cromosomi uguali od omologhi (ogni membro di una coppia di cromosomi omologhi deriva dal proprio padre ed uno dalla propria madre). Al termine, potremo osservare che sono presenti 22 coppie di cromosomi omologhi, numerate da 1 a 22, per un totale di 44 cromosomi (ad es.: due cromosomi 1, due cromosomi 2, due cromosomi 3, e così via fino a 22). I due rimanenti cromosomi, i cromosomi sessuali o “gonosomi”, sono di due tipi, detti “X” e “Y” per la somiglianza della loro forma a queste due lettere dell’alfabeto. Il cromosoma X è piuttosto grande, di dimensioni simili a quelle del cromosoma 6, mentre il cromosoma Y è molto piccolo, solitamente di dimensioni simili ad un cromosoma 21 o 22 e contiene pochi geni. Il referto definitivo lo si ottiene dopo circa 20 giorni dall'esecuzione del prelievo.
Anomalie Cromosomiche: Classificazione e Impatto
Negli individui sono possibili anomalie cromosomiche, numeriche o strutturali. Le anomalie numeriche implicano una variazione sul numero dei cromosomi e implicano problematiche di salute e di sviluppo.
Anomalie Cromosomiche Numeriche (Aneuploidie)
Le anomalie cromosomiche di numero più frequentemente riscontrate sono la trisomia 21 (Sindrome di Down), la trisomia 18 (Sindrome di Edwards), la trisomia 13 (Sindrome di Patau), tutte associate al ritardo mentale e, talora, a malformazioni e difetti di crescita.
- Sindrome di Down: Trisomia del cromosoma 21 (47, XX, +21 o 47, XY, +21).
- Sindrome di Patau: Trisomia del cromosoma 13 (47, XX, +13 o 47, XY, +13).
- Sindrome di Edwards: Trisomia del cromosoma 18 (47, XX, +18 o 47, XY, +18).
Anche i cromosomi del sesso possono andare incontro a difetti sia numerici (polisomie), ma spesso sono causa di una sintomatologia più lieve. Tra queste, le più note sono:
- Sindrome di Klinefelter: presenza di un cromosoma X in più (47, XXY).
- Sindrome di Turner: mancanza del cromosoma X nelle donne (45, X0).
- Altre polisomie: 47, XXX; 47, XYY.
Per quel che concerne le numerose alterazioni che si associano ai cromosomi sessuali (es. 45,X; 47,XXX; 47,XXY; ecc.), i genitori vanno informati che il più delle volte sono compatibili con un fenotipo perfettamente normale.
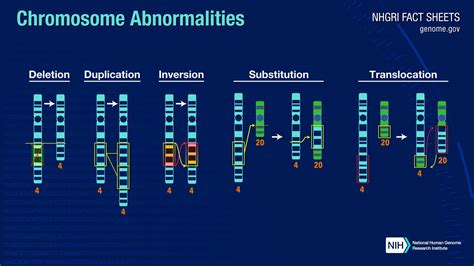
Anomalie Cromosomiche Strutturali
L'analisi del cariotipo permette anche di rilevare alterazioni della struttura cromosomica, come delezioni, inversioni e traslocazioni.
CARIOTIPO: POLIPLOIDIA e ANEUPLOIDIA
Le alterazioni cromosomiche strutturali bilanciate non danno luogo né a perdita né ad guadagno di materiale genetico e le persone portatrici sono generalmente fenotipicamente normali. Possono essere sia ereditate da un genitore (portatore sano) o possono verificarsi “de novo” e quindi essere riscontrate solo nelle cellule fetali. Le anomalie sbilanciate, invece, provocano perdita/guadano di materiale genetico, perciò vengono identificate in soggetti con fenotipo clinico.
- Delezioni: consistono nella perdita di un segmento di un cromosoma, che può essere terminale o interstiziale. Di solito le sindromi da delezione interessano segmenti relativamente grandi di cromosoma (> 10 Mb = Megabasi). Delezioni di queste dimensioni possono essere identificate con tecniche di citogenetica tradizionale.
- Inversioni: originano da 2 rotture che avvengono sullo stesso cromosoma e dalla successiva rotazione di 180° del segmento compreso tra i punti di rottura. Le inversioni producono un nuovo allineamento dei geni lungo l’asse di un cromosoma e di solito non si associano ad alterazioni cliniche.
- Traslocazioni reciproche: originano dalla rottura di due o, raramente, di più cromosomi e dallo scambio reciproco dei segmenti, senza perdita o acquisizione di materiale cromosomico. In questo caso si tratta di traslocazioni bilanciate, che hanno una frequenza di circa 1:1.000 nella popolazione generale.
- Traslocazioni Robertsoniane: hanno una frequenza di 1:1.000 nella popolazione ed originano dalla fusione di due cromosomi acrocentrici, che si sono rotti al centromero od in prossimità di questo. I cromosomi che si originano da questo scambio sono uno dotato di uno o due centromeri ed uno acentrico (cioè senza centromero). Quest’ultimo è instabile e viene perso alla prima divisione cellulare. I portatori di traslocazioni Robertsoniane hanno di conseguenza 45 cromosomi e sono clinicamente normali.
- Cromosomi ad anello (ring): sono originati dalla rottura di entrambe le braccia di un cromosoma, perdita delle regioni distali alle rotture e riunione delle due estremità in una struttura ad anello. I ring possono essere soprannumerari, ed in tal caso il portatore avrà 47 cromosomi e sarà trisomico per le regioni comprese nel ring, oppure possono aver sostituito un cromosoma normale, ed in tal caso il portatore avrà 46 cromosomi ma sarà parzialmente monosomico, per la perdita delle regioni distali alle rotture.
Complessità nell'Interpretazione dei Risultati
L’esame standard, che viene eseguito di routine in tutti i laboratori, mette in evidenza con chiarezza sia le aneuploidie, cioè le alterazioni del numero dei cromosomi, responsabili delle sindromi più frequenti (come ad esempio la trisomia 21 o Sindrome di Down), che le alterazioni strutturali di una certa entità. Ciò significa che sono evidenziabili con l’esame standard solo le anomalie strutturali più grandi di 10-15 Mb con una risoluzione di bandeggio di 350-400 bande.
Insuccesso della Coltura
È possibile talora che le cellule poste in coltura non crescano adeguatamente, in quanto sono presenti numerose cellule di tipo epiteliale di origine materna che impediscono la crescita anche minima. Si parla in questo caso di insuccesso della coltura. Questa evenienza è comunque rara (avviene in 1 caso su 500 nel caso di liquido amniotico; in 1 caso su 100 nel caso di villi coriali, nel nostro centro). È importante mettere in evidenza che la mancata crescita non è assolutamente indice di condizione patologica del feto.
Rischio Interpretativo e Artefatti
L’analisi citogenetica delle cellule del liquido amniotico riflette nel 99% dei casi il patrimonio cromosomico del feto ed identifica il corrispondente fenotipo, ma ci sono dei casi in cui traslocazioni o mosaicismi rendono difficile la definizione fenotipica. È possibile la presenza di artefatti “in vitro”, infatti, questo può avvenire dal 2% al 3% delle colture. Tra le altre si verificano più frequentemente poliploidie. Il cromosoma 20, per esempio, è uno dei cinque cromosomi più frequentemente coinvolti in pseudomosaicismi. L’origine delle cellule è probabilmente extraembrionaria ed il mosaicismo, in tali casi, non è ovviamente presente nei tessuti fetali.
La possibile discrepanza tra l’assetto cromosomico dei villi coriali e il cariotipo fetale con la possibilità di falsi positivi o falsi negativi è un aspetto da considerare. I falsi positivi (l’incidenza riportata in ampie casistiche è 1%) sono segnalati soprattutto quando viene utilizzata la sola tecnica diretta e sono controllabili sulla coltura o eventualmente sul liquido amniotico nel secondo trimestre. Il mosaicismo, ovvero la presenza di due linee cellulari con differente assetto cromosomico all’interno dello stesso individuo, è una condizione. Le cellule dei villi coriali presentano la caratteristica di essere portatrici di mosaicismi veri e propri che poi, al controllo, non sono presenti nei feti. Tale mosaicismo viene riscontrato nell’1% dei campioni prelevati. In caso di mosaicismo la cromosomopatia potrebbe coinvolgere il feto o essere confinata solamente agli annessi extra-embrionari; occorre perciò estendere l’indagine ad altri tessuti fetali (es. liquido amniotico o sangue) per chiarirne il significato clinico.
L'insorgenza di aberrazioni “in vitro” è un'altra eventualità; la maggior parte delle aberrazioni cromosomiche riscontrate nelle villocentesi sono da riferirsi a pseudomosaicismi. Con tale termine si intende la presenza di un cromosoma extranumerario presente solo nei villi ma del tutto assente nel feto. Questi, ovviamente, non hanno significato clinico. Per stabilire che si tratta di tale artefatto, il genetista esperto si basa essenzialmente sulle seguenti due considerazioni. La prima è che la cellula aberrante è solitamente unica quando ci si trova a leggere un allestimento diretto e, in coltura, l’alterazione interessa pertanto un unico clone di crescita. In tal modo nelle cellule coltivate l’aberrazione appartiene sempre a zone isolate di una stessa fiasca. La seconda considerazione è che, solitamente, la stessa alterazione non si riscontra nel feto.
Necessità di Approfondimenti Diagnostici di Secondo Livello
In alcuni casi si riscontrano anomalie cromosomiche particolari di cui non si conosce l’espressività fenotipica. Si tratta il più delle volte di piccoli porzioni cromosomiche soprannumerarie (markers), oppure anomalie cromosomiche strutturali come inversioni o traslocazioni, apparentemente bilanciate, che interessano essenzialmente gli autosomi. L’indagine sui genitori è di grande ausilio poiché, spesso, si riscontra la stessa anomalia in uno di essi. Qualora ci si trovasse, invece, di fronte ad una mutazione “de novo” avvenuta nel feto, è necessario un approfondimento diagnostico mediante tecnica array-CGH (cariotipo molecolare), al fine di stabilire se nella traslocazione o inversione vi sia stata perdita (delezione) o guadagno (duplicazione) di materiale genetico.
Il Cariotipo al di Fuori della Gravidanza: Fertilità e Altre Patologie
L'analisi del cariotipo non è limitata alla diagnosi prenatale, ma è un test genetico essenziale per valutare la fertilità sia nell’uomo che nella donna. La sua analisi permette di individuare eventuali alterazioni cromosomiche che possono influire sulla capacità riproduttiva o aumentare il rischio di aborti ripetuti. Per capire meglio quando è consigliabile eseguire questo test, come si svolge e cosa significano i suoi risultati, è fondamentale affidarsi a specialisti.

L’analisi del cariotipo è un test genetico essenziale per i pazienti che si rivolgono a una clinica di fertilità dopo aver incontrato difficoltà nel concepire naturalmente. In quanto tale, è considerata una valutazione di base nei test di fertilità femminile e maschile.
Indicazioni per il Cariotipo nella Valutazione della Fertilità
L'analisi del cariotipo è un test genetico consigliato in diverse situazioni per valutare la salute cromosomica di una persona. Può essere consigliato nei seguenti casi:
Se si sta pianificando una gravidanza:
- Per individuare se voi o il vostro partner avete alterazioni cromosomiche che potrebbero essere ereditate dal bambino.
- Per indagare sulle possibili cause di infertilità o aborti ricorrenti.
- Per valutare il rischio che il feto presenti anomalie genetiche, soprattutto in gravidanze con fattori di rischio quali: età materna avanzata (35 anni o più), poiché il rischio di alterazioni cromosomiche aumenta con l’età; storia familiare di malattia genetica o presenza di una malattia genetica in uno dei genitori; risultati anomali del test di screening prenatale.
Se si ha una storia familiare di disturbi cromosomici:
- Permette di valutare la possibilità di sviluppare una malattia genetica o di trasmetterla alle generazioni future.
Se voi o il vostro bambino avete i sintomi di una malattia genetica:
- A seconda dei sintomi e del tipo di disturbo, l’analisi del cariotipo può aiutare a confermare la diagnosi.
Se vi sono stati diagnosticati alcuni tipi di cancro o di malattie ematologiche:
- Alcuni tumori e malattie del sangue sono legati ad alterazioni cromosomiche che possono influenzare la diagnosi e la scelta del trattamento più appropriato. Tra questi vi sono: leucemia, linfoma, mieloma multiplo, anemia.
Il cariotipo è un esame essenziale per le coppie che cercano assistenza riproduttiva a causa di problemi di fertilità. Tuttavia, non tutte le cliniche lo richiedono di routine, ma di solito è raccomandato in situazioni specifiche come: azoospermia (assenza di spermatozoi nel liquido seminale); grave oligozoospermia (bassa concentrazione di spermatozoi); fallimento dell’impianto embrionale dopo trattamenti di riproduzione assistita; aborti spontanei ricorrenti, quando si verificano diverse perdite gestazionali consecutive. Se avete dubbi sulla necessità di questo test, è meglio consultare uno specialista della fertilità che possa guidarvi in base al vostro caso specifico.
Le donatrici vengono sottoposte al test di cariotipizzazione? Sì, l’analisi del cariotipo è un test essenziale prima che una persona possa essere accettata in un programma di donazione di gameti. Questo test permette di individuare eventuali anomalie cromosomiche che potrebbero influire sulla salute del futuro bambino. Nel caso in cui il candidato presenti alterazioni del cariotipo, non potrà partecipare al programma di donazione.
Cariotipo Alterato: Test e Trattamenti nella Riproduzione Assistita
Se lo studio del cariotipo rivela alterazioni cromosomiche, le tecniche di riproduzione assistita possono essere un’opzione per ottenere una gravidanza sana. In questi casi, si consiglia la fecondazione in vitro (FIVET) con diagnosi genetica preimpianto (DGP) o meglio conosciuta come PGT-A. Questa procedura consente di fecondare gli ovuli con gli spermatozoi in laboratorio e di analizzare geneticamente gli embrioni prima di trasferirli nell’utero, assicurando che vengano impiantati solo quelli con una dotazione cromosomica normale. Quelli con anomalie genetiche vengono scartati. Quando vengono rilevate alterazioni cromosomiche più complesse o gravi, la donazione di gameti può essere una valida alternativa, garantendo così maggiori possibilità di successo del trattamento.
Il Ruolo Cruciale della Consulenza Genetica
Importante è il counseling preconcezionale specie per le coppie a rischio riproduttivo, da richiedere ed eseguire prima del concepimento, quando la coppia o la donna desidera una gravidanza. A volte, infatti, nelle famiglie sono presenti malattie ereditarie, come cardiopatie, fibrosi cistica, sordità e così via, che potrebbero essere trasmesse al futuro nascituro.
È quindi comunque di fondamentale importanza il ruolo della consulenza genetica, che dovrebbe sempre accompagnare la diagnosi prenatale ed essere consigliata almeno in caso di referti che differiscano dal “cariotipo normale”. Questi esami devono essere sempre accompagnati dalla consulenza di un genetista, che spieghi alla coppia il significato dei risultati riportati sul referto, le conseguenze in termini di rischio e le possibili cure.
Interpretazione dei Risultati del Cariotipo
I risultati del cariotipo possono essere di due tipi principali:
- Risultato normale o negativo: indica che il test ha rilevato un totale di 46 cromosomi senza alterazioni strutturali o numeriche. Ciò significa che non sono state identificate anomalie cromosomiche.
- Risultato anormale o positivo: significa che sono state riscontrate alterazioni nel numero o nella struttura dei cromosomi. Queste alterazioni possono avere implicazioni diverse per la salute della persona o del bambino, a seconda del tipo di alterazione cromosomica individuata.
Se i risultati mostrano anomalie, è essenziale consultare uno specialista di genetica o di riproduzione assistita per comprenderne l’impatto e determinare i passi successivi da compiere. L’analisi del cariotipo fornisce informazioni sulla struttura e sul numero dei cromosomi, ma non rileva le mutazioni genetiche specifiche che causano le malattie monogeniche. In breve, la cariotipia è uno strumento fondamentale nella medicina riproduttiva e genetica, che aiuta a identificare i rischi e a prendere decisioni sulla pianificazione familiare.
tags: #valutazione #cariotipo #fetale #quando #farlo