Il panorama delle patologie genetiche è vasto e complesso, abbracciando condizioni che variano dalle anomalie del numero dei cromosomi a disordini del metabolismo enzimatico. Tra queste, la trisomia 18, nota anche come sindrome di Edwards, rappresenta una delle condizioni cromosomiche più gravi, caratterizzata dalla presenza di un cromosoma 18 in soprannumero. Parallelamente, nel campo delle alterazioni metaboliche ereditarie, si colloca la trimetilaminuria (TMAU), una condizione che, pur non essendo fatale, incide profondamente sulla qualità della vita sociale dei soggetti colpiti, manifestandosi con un odore corporeo peculiare.
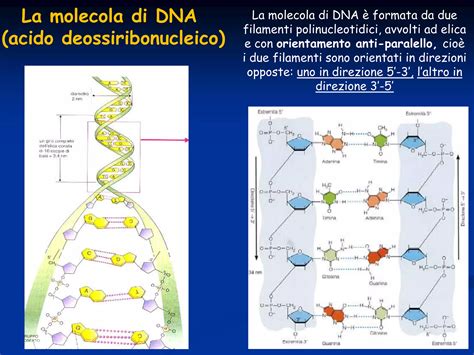
La Natura Biologica della Trisomia 18
La trisomia 18 è una malattia cromosomica causata da una copia supplementare del cromosoma 18. I cromosomi sono strutture presenti nelle cellule che contengono il DNA e svariati geni. I geni sono segmenti di acido desossiribonucleico (DNA) che contengono il codice per una specifica proteina che funziona in uno o più tipi di cellule dell’organismo. I geni contengono istruzioni che determinano l’aspetto e il funzionamento probabile dell’organismo. La presenza di un cromosoma supplementare, ossia 3 cromosomi dello stesso tipo invece dei normali 2, viene detta trisomia.
I bambini con trisomia 18 possiedono un terzo cromosoma 18. Il numero totale di cromosomi di un individuo affetto dalla sindrome di Edwards è quindi 47 e non 46. La trisomia 18 si osserva in circa 4 gravidanze su 10.000, con un'incidenza stimata in circa 1/6000-1/8000 nati. Come per la gran parte delle anomalie cromosomiche numeriche, il meccanismo che porta alla trisomia è una “non disgiunzione meiotica”, quasi sempre di derivazione materna, che ha luogo prima del concepimento e che riguarda una cellula sessuale di uno dei genitori, oppure poco dopo il concepimento. Le madri di oltre 35 anni di età presentano un rischio maggiore di avere un bambino con trisomia 18.
Varianti della Condizione Cromosomica
Non tutti i casi di trisomia 18 si presentano con la stessa configurazione genetica. Esistono diverse forme:
- Forma completa: Circa il 94% dei bambini con sindrome di Edwards ha la forma completa, cioè ogni cellula del corpo possiede tre copie del cromosoma 18, invece di due.
- Trisomia a mosaico: Circa il 5% dei bambini ha la copia extra del cromosoma 18 solo in alcune cellule del corpo. Questa forma meno grave della malattia dipende dal tipo e dal numero di cellule che hanno il cromosoma supplementare. Alcuni bambini possono essere colpiti solo lievemente, mentre altri lo sono in modo grave.
- Forma parziale: In questa forma, la meno frequente, ad essere triplicato è solo un segmento del cromosoma 18. Il fenotipo è dovuto alla presenza di tre copie della regione 18q11-q12.
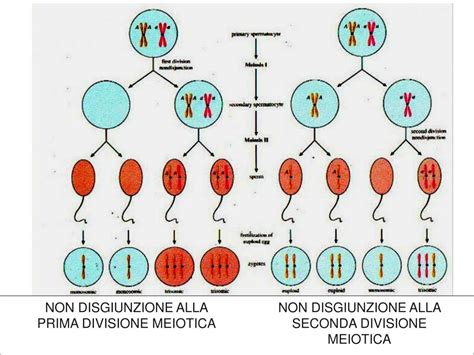
Quadro Clinico e Segni Distintivi della Trisomia 18
La trisomia 18 è una condizione molto grave e spesso incompatibile con la vita: la maggior parte di chi ne è affetto muore prima della nascita o nasce morto. Oltre il 95% dei feti colpiti muore in utero. I neonati e i bambini che sopravvivono alla gravidanza mostrano una lunga serie di sintomi e segni dagli effetti drammatici.
Anomalie Fisiche alla Nascita
Nell’utero, i feti colpiti di solito non sono molto attivi e spesso si osservano un eccesso di liquido amniotico (polidramnios), una placenta piccola e una crescita limitata. Alla nascita, i neonati presentano spesso un basso peso, ipotrofia e muscoli e grasso corporeo sottosviluppati. Appaiono fiacchi e hanno un pianto debole. Tra le caratteristiche cliniche comuni si annoverano la microcefalia con cranio stretto e dolicocefalia, la microretrognazia (mascella piccola), l'ipertelorismo e orecchie angolate a disegno semplice e in posizione bassa.
Le mani sono chiuse a pugno, con il dito indice che spesso si sovrappone al medio e all’anulare. Le unghie sono sottosviluppate. Le anomalie dei piedi comprendono il piede equino-varo e/o a piccozza. Sono comuni pieghe cutanee, specialmente sulla nuca, pelvi stretta e sterno corto.
Coinvolgimento degli Organi Interni
Anche gli organi interni presentano difetti gravi. Sono coinvolti quasi tutti gli apparati:
- Apparato cardiaco: Presente in oltre il 90% dei casi.
- Apparato gastrointestinale: Atresia esofagea, malformazioni ano-rettali, ernie e muscoli separati dalla parete addominale.
- Apparato genito-urinario: Idronefrosi, agenesia mono-bilaterale e, nei maschi, ritenzione dei testicoli.
- Sistema neurologico e polmonare: Nelle prime settimane sono presenti ipotonia, iporeattività e difficoltà alimentari (scarsa suzione), seguite spesso da una progressione verso l'ipertono.
Strumenti Diagnostici e Gestione
Prima della nascita, la trisomia 18 può essere sospettata in base ai risultati di un’ecografia del feto che evidenzia ritardo di crescita, malformazioni o cisti dei plessi corioidei. I medici eseguono screening prenatali, come il test combinato (bi-test e translucenza nucale) tra l'11a e la 14a settimana, o il NIPS (analisi del DNA fetale libero circolante nel sangue materno) dalla 10a settimana in avanti.
Per confermare la diagnosi, si utilizzano tecniche invasive:
- Villocentesi: Prelievo di tessuto trofoblastico intorno alla 10a-11a settimana.
- Amniocentesi: Prelievo di liquido amniotico intorno alla 16a-18a settimana, considerato l'esame definitivo e più attendibile.
Dopo la nascita, l’aspetto fisico può suggerire la diagnosi, che viene confermata mediante l’analisi del cariotipo (esame del sangue). Non esiste una cura per la trisomia 18; la presa in carico è puramente di supporto. Sebbene il trattamento chirurgico delle malformazioni possa essere tentato, esso migliora solo minimamente la prognosi. Il 90% dei bambini muore nel primo anno di vita. Tuttavia, grazie a cure più attente, alcuni soggetti raggiungono l'età adulta, presentando però un rischio aumentato di sviluppare tumori come l'epatoblastoma e il tumore di Wilms, rendendo necessari esami periodici di diagnostica per immagini.
Test del DNA fetale: cos'è e perché farlo (I° trimestre)
La Trimetilaminuria: La Sindrome da Odore di Pesce
Se la trisomia 18 è un'alterazione della struttura cromosomica numerica, la trimetilaminuria (TMAU) rappresenta un disordine metabolico differente, spesso definito "sindrome da odore di pesce". È un difetto genetico ereditario dovuto a mutazioni a carico del gene FMO3, che comporta l’emissione di un forte e sgradevole odore paragonabile a quello del pesce marcio. Questa sostanza, la trimetilammina (TMA), non viene correttamente metabolizzata dall'organismo e viene rilasciata attraverso sudore, urine e respiro.
Impatto Sociale e Diagnosi
Dal punto di vista medico, la TMAU non comporta rischi vitali, ma è estremamente invalidante sotto il profilo sociale. Chi ne è affetto può subire isolamento, mobbing lavorativo e sviluppare patologie psichiatriche come depressione e ansia. La comparsa della patologia è legata a varianti genetiche trasmesse dai genitori, ma può manifestarsi anche in correlazione a malattie croniche del fegato o alterazioni della flora intestinale.
La diagnosi avviene attraverso il dosaggio della TMA nelle urine e viene confermata mediante test genetico. Attualmente, l’unico strumento per tenere sotto controllo l’odore consiste nella dietoterapia: una dieta drastica che limita il consumo di alimenti ricchi di colina, L-carnitina e lecitina (presenti in pesce, crostacei, uova e fegato). Il percorso richiede il supporto di un'equipe medica dedicata alle malattie metaboliche.
Confronto tra Anomalie Cromosomiche e Delezioni
È importante distinguere le trisomie da altre anomalie cromosomiche come le delezioni e le duplicazioni. Mentre la trisomia implica la presenza di un cromosoma in più, le delezioni consistono nell'assenza di un tratto di cromosoma, con conseguente perdita dei geni ivi localizzati. Esempi includono la sindrome del cri-du-chat (delezione sul cromosoma 5) o la delezione del braccio corto del cromosoma 1 (1p36). Queste condizioni causano dismorfismi facciali, ritardo mentale, anomalie cardiache e disturbi dell'alimentazione, confermando che la stabilità del corredo cromosomico è essenziale per il normale sviluppo neuro-cognitivo e fisico dell'essere umano.
La gestione di tutte queste condizioni, che si tratti di un'anomalia cromosomica severa o di un disordine metabolico cronico, richiede non solo competenze mediche avanzate, ma anche una profonda comprensione del vissuto psicologico dei pazienti e delle loro famiglie, per garantire un supporto che vada oltre la semplice diagnosi genetica.