Introduzione: La Fibrosi Cistica, una Patologia Ereditaria Complessa
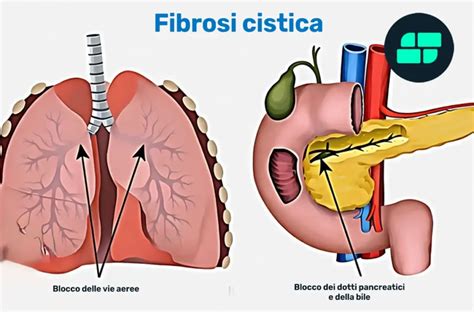
La fibrosi cistica (FC), conosciuta anche come mucoviscidosi, è una delle malattie genetiche ereditarie gravi più diffuse, sebbene sia classificata come rara in base alla sua incidenza. Questa patologia colpisce indifferentemente maschi e femmine e può manifestarsi fin dalla nascita. A causare la malattia è un difetto della proteina CFTR (Cystic Fibrosis Transmembrane Conductance Regulator), la cui funzione cruciale è quella di regolare gli scambi idroelettrolitici, in particolare il corretto passaggio di cloro e sodio, attraverso le membrane cellulari. Quando questa proteina è alterata, si verifica un'anomalia nel trasporto di sali, che determina la produzione di secrezioni "disidratate". Ciò significa che il sudore diventa molto ricco di sodio e cloro, mentre il muco, particolarmente nelle vie aeree, diventa denso e vischioso, tendendo a ostruire i dotti nei quali è chiamato a scorrere.
Gli effetti di questa disfunzione si ripercuotono su diversi organi e apparati dell'organismo. Le persone con FC sono colpite soprattutto nell'apparato respiratorio, incluse le vie aeree, i bronchi e i polmoni, dove il ristagno di secrezioni dense favorisce infezioni respiratorie recidivanti che possono diventare croniche. Altre aree significativamente coinvolte sono il pancreas, con conseguente malfunzionamento esocrino e problemi di digestione e malnutrizione, il fegato, l'intestino e l'apparato riproduttivo, specialmente negli uomini, dove può causare infertilità a causa dell'ostruzione dei dotti spermatici. La comprensione approfondita di questa patologia è fondamentale, soprattutto alla luce delle nuove frontiere terapeutiche che si aprono, in particolare per i neonati e i bambini. In Italia, si stima che su ogni 2.500-3.000 bambini nati, uno sia affetto da fibrosi cistica, con quasi 6.000 bambini, adolescenti e adulti affetti curati nei centri specializzati. Questa frequenza relativamente bassa, ma non trascurabile, ha portato a un'attenzione particolare nel nostro Paese, culminata anche in una legge specifica, la 548/93, dedicata alla prevenzione e alla cura della fibrosi cistica, che le ha conferito uno status unico al di fuori dell'elenco delle patologie rare esenti.
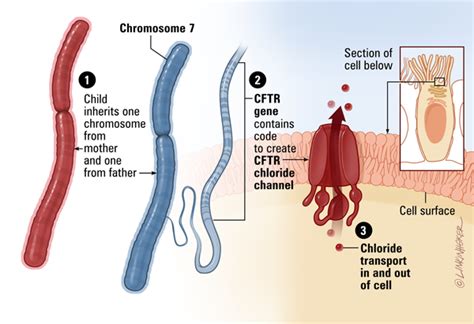
Le Cause Genetiche e la Trasmissione della Fibrosi Cistica
La fibrosi cistica è una malattia genetica autosomica recessiva, il che implica specifiche modalità di trasmissione ereditaria. Questo significa che la condizione si manifesta solamente se un individuo eredita due copie mutate del gene CFTR, una da ciascun genitore. Una persona che possiede una copia normale e una copia mutata del gene della fibrosi cistica è definita "portatore". Il portatore non mostra alcun sintomo della malattia, rimanendo perfettamente sano, ma è in grado di trasmettere la mutazione ai propri figli. L'identificazione di un portatore avviene generalmente a seguito della nascita di un figlio affetto o tramite uno specifico test genetico.
La frequenza dei portatori nella popolazione varia notevolmente. In Italia, si stima che circa una persona su 30 sia un portatore di fibrosi cistica. Di conseguenza, la probabilità che una coppia sia composta da due portatori si attesta attorno a 1 su 900. Quando una coppia di portatori intraprende una gravidanza, le probabilità di trasmissione della malattia seguono precise leggi mendeliane:
- Una probabilità su quattro (25%) di avere un figlio affetto da Fibrosi Cistica.
- Una probabilità su due (50%) di avere un figlio portatore, ma non malato.
- Una probabilità su quattro (25%) di avere un figlio né portatore né malato.
Queste percentuali evidenziano l'importanza dei test genetici, specialmente per le coppie che pianificano una gravidanza o che hanno parenti affetti dalla malattia. Il Servizio Sanitario Nazionale, ad oggi, offre il test per l'identificazione del portatore a persone con consanguinei affetti da Fibrosi Cistica e, in alcune circostanze, a coppie che intraprendono percorsi di procreazione assistita. Questo esame, che solitamente si effettua con un semplice prelievo del sangue, può essere proposto ai singoli o alla coppia in fase pre-concezionale o prenatale, durante la gravidanza, permettendo di capire se si è portatori sani e, quindi, in grado di trasmettere la mutazione ai figli. È importante sottolineare che anche una persona senza casi di malattia in famiglia può essere portatrice. La frequenza dei portatori mostra variazioni significative tra le diverse popolazioni, risultando maggiore in quelle europee e nordamericane rispetto a quelle di origine africana o asiatica. Tuttavia, è fondamentale riconoscere che gli attuali test di screening per la popolazione generale non sono perfetti, in quanto non riescono a individuare il 100% dei portatori. Dato che non tutte le circa 2.000 differenti mutazioni del gene CFTR sono conosciute e ricercate, alcuni soggetti potrebbero ottenere un risultato negativo al test pur essendo, in realtà, portatori di fibrosi cistica. Per chi desidera informarsi prima del concepimento, esistono test genetici pre-concepimento completi e non invasivi, come Genescreen® di Eurofins Genoma, che analizzano simultaneamente centinaia di malattie ereditarie, inclusa la fibrosi cistica.
Riconoscere la Fibrosi Cistica: Sintomi e Diagnosi Precoce
La fibrosi cistica si manifesta con una varietà di sintomi e gradi di gravità, che possono rendere la diagnosi complessa in assenza di screening sistematici. Tuttavia, la maggior parte dei casi oggi viene identificata molto precocemente, già nei primi giorni di vita, grazie all'introduzione dello screening neonatale obbligatorio in tutte le regioni italiane.
I sintomi tipici della fibrosi cistica sono il risultato diretto dell'anomalia nel trasporto di sali che causa secrezioni dense e vischiose. Questi includono:
- Apparato respiratorio: Infezioni respiratorie frequenti e recidivanti, spesso a causa del ristagno di muco nei bronchi e nei polmoni, che tendono a cronicizzare e favoriscono la proliferazione batterica. I pazienti possono presentare affanno e tosse persistente.
- Apparato digerente: Cattiva digestione e malnutrizione sono comuni a causa del malfunzionamento del pancreas esocrino, che ostruito dal muco denso non produce a sufficienza gli enzimi digestivi. Ciò può portare a un ritardo di crescita nel bambino. Sintomi gastrointestinali possono includere diarrea cronica.
- Altre manifestazioni: Un aumento della concentrazione di sale nel sudore è un segno distintivo della malattia. Si possono riscontrare anche epatopatia cronica e diabete. Negli uomini, l'ostruzione dei dotti spermatici può causare infertilità.
- Nei neonati: Oltre ai problemi respiratori e intestinali, si osserva uno scarso assorbimento dei nutrienti. In casi rari, alla nascita può manifestarsi l'ileo da meconio, un blocco intestinale causato da meconio (le prime feci del neonato) insolitamente denso.
La diagnosi di fibrosi cistica nei neonati si basa su una serie di test sicuri e affidabili che consentono di intervenire prima ancora che i sintomi si manifestino o si aggravino.
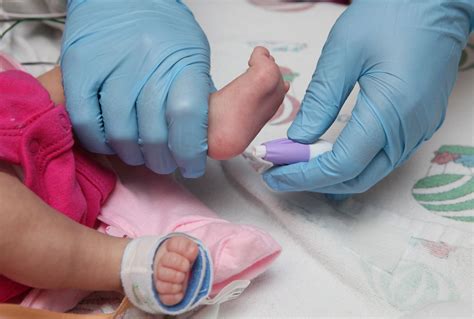
Lo screening neonatale: Questo è un test semplice e rapido, eseguito entro le prime 48-72 ore di vita del neonato mediante il prelievo di alcune gocce di sangue dal tallone. Le gocce vengono analizzate per rilevare la presenza di sostanze anomale, come la tripsina immunoreattiva (IRT). Livelli elevati di IRT possono indicare un rischio aumentato di fibrosi cistica. È fondamentale comprendere che un risultato positivo allo screening non costituisce una diagnosi definitiva, ma segnala la necessità di ulteriori accertamenti. Se il test rileva livelli di tripsina elevati, il Centro Regionale di Riferimento per gli Screening richiama la famiglia per un secondo dosaggio, tra il 15° e il 20° giorno di vita del bambino, per confermare l'indicazione. Grazie a questa procedura, ogni anno decine di bambini vengono identificati precocemente, permettendo un rapido accesso alle cure necessarie.
Il test del sudore: Questo esame è considerato da decenni il test di riferimento per la conferma della fibrosi cistica dopo uno screening neonatale positivo o in presenza di sintomi sospetti. Si tratta di un test non invasivo e indolore, che si esegue in ambulatorio e misura la concentrazione di cloro presente nel sudore. Un valore di cloro superiore a 60 mEq/L è generalmente considerato positivo e conferma la diagnosi di FC, mentre valori inferiori a 40 mEq/L sono considerati negativi. Per i valori che rientrano tra questi due range, il test può essere ripetuto per ottenere una diagnosi accertata. Il test del sudore è richiesto non solo in caso di screening neonatale positivo, ma anche per i bambini che hanno presentato alla nascita ileo da meconio, per quelli con sintomi respiratori o gastrointestinali che inducano a sospettare la diagnosi di FC, e in casi di pancreatite cronica o grave disidratazione, specialmente nei mesi estivi.
L'analisi del gene CFTR: Quando è necessaria maggiore precisione, l'analisi del gene CFTR, responsabile della regolazione del passaggio di cloro e sodio nelle cellule, consente di confermare la diagnosi e di individuare la tipologia specifica di mutazione. Conoscere la mutazione è diventato cruciale per l'accesso a terapie mirate. Ricevere una diagnosi di fibrosi cistica nelle prime settimane di vita può essere un momento di grande preoccupazione per i genitori, ma rappresenta in realtà un vantaggio fondamentale. Oggi, la medicina di precisione permette di personalizzare i trattamenti in base al profilo genetico del piccolo paziente, rendendo la gestione della malattia sempre più efficace e "su misura". Una diagnosi tempestiva non è quindi solo una notizia da affrontare, ma il punto di partenza di un percorso di cura e di fiducia nel futuro, con l'obiettivo di offrire ai neonati una vita lunga e serena.
Pediatria, arriva il test del sudore per la fibrosi cistica
L'Evoluzione delle Terapie Convenzionali e l'Aumento dell'Aspettativa di Vita
Fino a pochi decenni fa, la fibrosi cistica era considerata una malattia con una prognosi estremamente severa, con un'aspettativa di vita che, negli anni '60, raramente superava la prima decade. Tuttavia, grazie ai continui progressi nell'ambito delle cure e dell'assistenza, la percezione della malattia è significativamente cambiata: da patologia letale è diventata una condizione con la quale si può diventare adulti.
Gli standard di cura internazionali, unitamente all'introduzione capillare dello screening neonatale e a test diagnostici più sofisticati, hanno permesso di innalzare notevolmente l'aspettativa di vita delle persone affette da Fibrosi Cistica. Oggi, oltre il 50% della popolazione con FC in Italia è costituita da adulti, e l'aspettativa di vita si aggira attorno ai 40 anni, un dato in costante miglioramento. Questo progresso è attribuibile a una combinazione di fattori, tra cui la diagnosi precoce, l'istituzione di centri di cura dedicati e l'avanzamento delle terapie.
I trattamenti attualmente disponibili si raggruppano in diverse categorie, tutte orientate a gestire i sintomi della malattia e a prevenirne le complicanze, sebbene non esista ancora una terapia risolutiva unica e definitiva.
1. Terapia Farmacologica:
- Antibiotici: Le infezioni polmonari, favorite dal muco vischioso che ostruisce le vie respiratorie e crea un ambiente propizio alla proliferazione batterica, sono trattate con antibiotici somministrati per via orale o intravenosa. L'antibioticoterapia per via aerosolica riveste un ruolo importante nel controllo delle infezioni e dovrebbe essere prescritta in base al risultato dell'esame microbiologico sulle secrezioni bronchiali, ottenute dopo la tosse o mediante aspirato faringeo.
- Farmaci fluidificanti: Aerosol di antibiotici e farmaci specifici sono utilizzati per fluidificare le secrezioni, rendendo il muco meno denso e più facile da espellere.
2. Fisioterapia Respiratoria:
- La fisioterapia respiratoria quotidiana è un pilastro fondamentale del trattamento. Ha lo scopo di mantenere le vie aeree libere dal muco, prevenendo il ristagno e, di conseguenza, le infezioni respiratorie.
3. Nutrizione e Integrazione Enzimatica:
- A causa del malfunzionamento del pancreas, che compromette la digestione e l'assorbimento dei nutrienti, ai pazienti viene prescritta una nutrizione ipercalorica e un'integrazione enzimatica pancreatica. Gli enzimi digestivi sono somministrati sotto forma di capsule o polveri da assumere con i pasti. La quantità deve essere valutata tenendo conto della gravità della compromissione pancreatica e delle abitudini alimentari del paziente, che deve seguire una dieta libera, equilibrata, ipercalorica.
4. Farmaci Modulatori della Proteina CFTR:
- Negli ultimi anni, l'introduzione dei farmaci modulatori CFTR ha rappresentato una vera rivoluzione terapeutica. Questi farmaci agiscono direttamente sul difetto genetico, essendo in grado di correggere e/o potenziare il funzionamento della proteina CFTR difettosa. Hanno dimostrato di essere efficaci nel ridurre i sintomi polmonari e altri sintomi correlati alla fibrosi cistica, modificando significativamente il decorso della malattia. Tra questi, Ivacaftor ha ricevuto l'approvazione per pazienti con la mutazione G551D nel gene CTFR di età superiore ai 6 anni, pur non avendo mostrato efficacia per altre mutazioni come la R117H.
- Nonostante i loro benefici, i farmaci modulatori non coprono tutte le mutazioni del gene e sono indicati solo per specifiche fasce di età. In Italia, circa il 30% delle persone con Fibrosi Cistica rimane ancora escluso da queste cure innovative. Gli effetti collaterali a lungo termine non sono ancora completamente conosciuti, data la loro recente introduzione.
5. Trapianto Polmonare:
- Nei casi di insufficienza respiratoria grave e irreversibile, quando le terapie conservative non sono più sufficienti, il trapianto polmonare può rappresentare l'ultima opzione terapeutica disponibile per migliorare la qualità di vita e prolungare la sopravvivenza.
Dato che la fibrosi cistica è una malattia complessa che colpisce più organi e ha un impatto significativo sulla qualità della vita, la legge 548/93 ha istituito Centri multiprofessionali dedicati per curare al meglio le persone affette. Questi centri garantiscono una presa in carico continua, con controlli periodici e terapie personalizzate, monitoraggi respiratori e supporto nutrizionale, permettendo di intervenire tempestivamente su ogni variazione clinica. L'arrivo dei modulatori CFTR di ultima generazione ha inaugurato una nuova fase terapeutica, e accanto ai progressi farmacologici, si stanno diffondendo programmi di monitoraggio domiciliare e strumenti digitali che permettono alle famiglie di seguire in tempo reale l'andamento della malattia. Le complicanze, sebbene la sopravvivenza sia aumentata, continuano ad avere un effetto negativo sulla qualità della vita, sul peso psicologico e sui costi della terapia. L'obiettivo oggi non è più solo convivere con la fibrosi cistica, ma vivere bene, a lungo e con fiducia nel futuro.
La Promessa della Terapia Genica: Dal Concetto alla Ricerca Avanzata
La scoperta del gene CFTR nel 1989 suscitò un enorme clamore all'epoca, alimentando grandi speranze riguardo alla possibilità di una futura terapia genica. Il concetto alla base della terapia genica è affascinante e in linea di principio relativamente lineare: introdurre nelle cellule di un paziente copie normali di geni, allo scopo di correggere una malattia genetica causata da geni mutanti o difettosi. In questo modo, le cellule malate verrebbero, teoricamente, "guarite". Questo approccio terapeutico fu subito proposto come assai logico, particolarmente per le malattie ereditarie mendeliane (o monogeniche) come la fibrosi cistica.
Tuttavia, il passaggio dalla teoria alla pratica si è dimostrato un percorso lungo e arduo. Sono stati necessari circa trent'anni perché la terapia genica diventasse una realtà clinica consolidata, come lo è oggi per i bambini con certi tipi di immunodeficienza grave o, più recentemente, per l'emofilia B. Per la fibrosi cistica, le sfide sono state particolarmente complesse, soprattutto a causa della necessità di correggere un numero sufficiente di cellule nell'epitelio respiratorio, un organo vasto e costantemente esposto ad agenti esterni.
Per realizzare l'obiettivo della terapia genica, sono necessari due elementi chiave:
- A) Un trasportatore o vettore efficiente: Questo vettore ha il compito di veicolare il gene normale all'interno delle cellule bersaglio. I vettori più efficienti si sono rivelati essere alcuni virus, opportunamente modificati e resi innocui per l'uomo. Tuttavia, poiché l'epitelio respiratorio è direttamente accessibile dall'esterno, la ricerca ha esplorato anche l'uso di vettori non virali, come particelle inalate tramite aerosol.
- B) Una profonda comprensione del funzionamento del gene CFTR: È essenziale sapere come il gene CFTR è regolato e come la sua proteina esercita la sua funzione per poterlo sostituire o correggere efficacemente. Le conoscenze in questo campo sono notevolmente avanzate negli anni.
In modelli di topo e anche in pazienti con FC, sono stati compiuti progressi significativi sia nella messa a punto dei vettori (A) sia nella comprensione del gene (B). Purtroppo, finora, questi studi non hanno prodotto risultati clinici apprezzabili. La ragione principale è di carattere quantitativo: anche se fosse possibile correggere il difetto genetico in una percentuale relativamente piccola, diciamo il 5%, delle cellule dell'epitelio respiratorio, questo potrebbe risultare insufficiente dal punto di vista clinico per ottenere un beneficio tangibile per il paziente. L'impedimento principale è stato quello di riuscire a trattare un numero sufficientemente elevato di cellule in modo duraturo.

Questo limite ha spinto la ricerca a esplorare approcci più innovativi, che potrebbero finalmente permettere alla terapia genica per la fibrosi cistica di superare la fase sperimentale per entrare nella pratica clinica con risultati significativi. Tra queste frontiere, emerge con particolare forza l'idea di utilizzare le cellule staminali, come discusso nella prossima sezione.
Frontiere Innovative: Il Ruolo delle Cellule Staminali e dei Vettori Lentivirali
Se finora l'ostacolo maggiore alla terapia genica per la fibrosi cistica è stato di natura quantitativa, ovvero la difficoltà di correggere un numero sufficiente di cellule in modo efficace e duraturo, la ricerca sta ora affrontando questa sfida in un modo nuovo e promettente. Il prossimo protocollo di terapia genica per la FC potrebbe prevedere un approccio basato sulle cellule staminali e sull'uso di vettori lentivirali.
Il protocollo che sta emergendo dalla ricerca prevede i seguenti passaggi:
- Isolamento delle cellule staminali: Attraverso una biopsia, si prelevano cellule staminali dall'epitelio respiratorio del paziente affetto da fibrosi cistica. Le cellule basali delle vie aeree, in particolare, sono cellule staminali che fungono da progenitrici per tutte le altre cellule dell'epitelio respiratorio.
- Correzione genetica in vitro: Le cellule staminali isolate vengono coltivate in laboratorio. In questa fase, il gene CFTR normale viene trasferito nelle cellule, probabilmente tramite un vettore lentivirale.
- Verifica e selezione: Si verifica che il gene sia stato trasferito con successo e che si esprima correttamente all'interno delle cellule. Solo le cellule che hanno incorporato e attivano il gene corretto vengono selezionate.
- Reintroduzione nel paziente: Le cellule staminali geneticamente corrette vengono reintrodotte nel paziente per via endobronchiale. L'aspettativa è che queste cellule possano ora colonizzare l'epitelio dell'albero respiratorio.
Questo approccio rappresenta una forma di rigenerazione cellulare. L'idea è che, a partire da quelle poche cellule staminali sulle quali è stata eseguita la correzione genetica in vitro, si possano generare in vivo milioni di nuove cellule epiteliali mature, tutte contenenti la copia funzionante del gene CFTR. Questo processo di rigenerazione spesso avviene spontaneamente quando il CFTR è funzionante ma l'epitelio è danneggiato, una condizione fin troppo frequente nei pazienti con FC. L'integrazione permanente del gene corretto nelle cellule basali, che sono cellule staminali con la capacità di replicarsi e differenziarsi, potrebbe potenzialmente produrre benefici all'organismo per tutta la vita del paziente, poiché le nuove cellule prodotte sarebbero geneticamente corrette.
I vettori lentivirali (LV) sono considerati uno dei candidati principali per la terapia genica mirata alle vie aeree colpite dalla fibrosi cistica. La loro capacità di integrare in modo permanente il gene desiderato nel genoma delle cellule ospiti li rende particolarmente adatti per questo scopo, promettendo un effetto terapeutico a lungo termine. Sviluppare una terapia genica che sia in grado di fornire in modo efficiente una copia corretta di CFTR, migliorando così la salute dei polmoni, è l'obiettivo primario della ricerca in questo ambito.
Tuttavia, i vettori lentivirali devono affrontare una sfida significativa: per avere un effetto terapeutico, devono essere trasportati efficacemente attraverso le vie aeree superiori, dove iniziano le problematiche causate dalla fibrosi cistica. Per superare questo ostacolo, i ricercatori hanno optato per l'utilizzo di particelle magnetiche coniugate ai vettori, una tecnica nota come magnetofezione.
La magnetofezione è un metodo in cui un campo magnetico viene utilizzato come agente di trasfezione per migliorare il trasferimento del DNA alle cellule. Questa tecnica è comunemente impiegata in vitro per linee cellulari difficili da trasdurre, sia con vettori non virali che virali. L'efficacia della magnetofezione con LV è stata dimostrata in studi precedenti su cellule umane in vitro, inclusi modelli di fibrosi cistica. L'applicazione di questa tecnica in vivo, tuttavia, ha ricevuto relativamente poca attenzione ed è stata valutata solo in un numero limitato di studi su modelli animali, con un numero ancora minore di studi specifici sui polmoni.
In uno studio recente, è stata utilizzata una particolare tecnica di imaging, definita "a contrasto di fase", per visualizzare in modo non invasivo il movimento in vivo di magnetoparticelle nella trachea di ratti vivi. Questo ha permesso di comprendere meglio il funzionamento della tecnologia in vivo, ottenendo informazioni utili per la sua ottimizzazione. È stato evidenziato che le particelle non potevano essere facilmente trascinate con il magnete lungo la superficie delle vie aeree, ma durante la somministrazione la deposizione si concentrava dove il campo magnetico era più forte. Questa ricerca suggerisce che, pur essendoci ancora ostacoli, l'uso combinato di cellule staminali, vettori lentivirali e tecniche di veicolazione avanzate come la magnetofezione potrebbe aprire nuove e concrete possibilità per una terapia genica efficace e duratura per la fibrosi cistica.
Oltre l'Addizione Genica: La Correzione Diretta e l'Editing Molecolare
L'introduzione dei farmaci modulatori, come Ivacaftor, ha evidenziato in modo tangibile come pazienti affetti da fibrosi cistica con mutazioni diverse richiedano terapie altrettanto diverse e personalizzate. Allo stesso modo, la terapia genica fin qui prospettata, basata sull'aggiunta di un gene normale, potrebbe applicarsi a tutti i pazienti le cui mutazioni causano una mancata produzione della proteina CFTR o la produzione di una proteina non funzionante. Tuttavia, esiste una considerazione cruciale per alcune tipologie di mutazioni.
In certi casi, la proteina anormale prodotta dal gene mutante potrebbe interferire con la localizzazione o con la funzione della proteina normale prodotta dal gene che è stato introdotto con la terapia genica additiva. Questo fenomeno, noto come effetto "dominante negativo", potrebbe vanificare o ridurre l'efficacia dell'intervento. In tali circostanze, un approccio ancora più promettente e auspicabile rispetto alla semplice "addizione" del gene normale sarebbe la sostituzione del gene mutato (o della sua parte mutata) con l'omologo normale. Questo è il principio su cui si basa la correzione genetica diretta, o più ampiamente, il concetto di editing molecolare.
L'editing molecolare, come nuova frontiera della terapia genica, prevede la correzione diretta del gene difettoso anziché la sua semplice integrazione di una copia normale. Questa tecnologia avanzata consente di "tagliare e incollare" segmenti specifici del DNA, rimuovendo la porzione mutata e inserendo quella corretta. Questo approccio offre il vantaggio di non lasciare "tracce" del gene mutato e di ripristinare la funzione del gene nel suo contesto genomico naturale, evitando potenziali interferenze con le proteine endogene. Un esempio di successo di correzione genetica diretta è stato dimostrato nell'anemia falciforme mediante l'utilizzo di nucleasi a dita di zinco ingegnerizzate in cellule staminali pluripotenti indotte umane.
La realizzazione pratica di un protocollo terapeutico così avanzato, basato sulla correzione genetica in vivo o ex vivo con reintroduzione di cellule staminali, non sarà certamente facile. Le complessità tecniche, etiche e regolatorie sono notevoli. Tuttavia, le tecnologie di trasferimento genico e di ricombinazione omologa, che sono alla base di queste tecniche di editing, hanno progredito enormemente rispetto al 1989, quando il gene CFTR fu identificato per la prima volta. La ricerca è in fermento a livello globale: in Gran Bretagna, ad esempio, esiste già un consorzio con l'obiettivo preciso e specifico di sviluppare la terapia genica per la fibrosi cistica. Questo impegno collettivo, unito all'avanzamento delle conoscenze e delle tecnologie, alimenta la speranza che la fibrosi cistica, una malattia così impattante, possa un giorno essere trattata alla radice con una terapia definitiva.
L'Importanza dello Screening del Portatore e le Implicazioni Socio-Etiche
La gestione della fibrosi cistica non si limita alla diagnosi precoce e al trattamento dei pazienti, ma include anche una fondamentale dimensione di prevenzione e consapevolezza per la popolazione generale. In questo contesto, lo screening del portatore di fibrosi cistica assume un'importanza cruciale. Per valutare l'impatto complessivo di un eventuale screening del portatore esteso a individui e coppie in età fertile, indipendentemente dal grado di familiarità con la malattia, l'Istituto Mario Negri, in collaborazione con la LIUC - Università Cattaneo, ha condotto una valutazione di Health Technology Assessment (HTA).
L'obiettivo di questo studio multidisciplinare era analizzare l'impatto economico-organizzativo, sociale, etico, nonché gli aspetti di sicurezza ed efficacia derivanti dall'introduzione di un tale programma di screening. Il progetto, finanziato dalla Fondazione per la Ricerca sulla Fibrosi Cistica e con la responsabilità scientifica di Carlo Castellani, responsabile del Centro Fibrosi Cistica dell’Ospedale IRCCS Istituto Giannina Gaslini di Genova, ha impiegato diverse metodologie per condurre la valutazione delle dimensioni di interesse, tra cui la revisione della letteratura, modelli di analisi economica e interviste qualitative.

I risultati della valutazione di Health Technology Assessment hanno evidenziato punti importanti. Dalla revisione sistematica della letteratura è emersa una mancanza di dati solidi e aggiornati sull'efficacia a lungo termine di uno screening del portatore esteso a tutta la popolazione. Tuttavia, i modelli di valutazione economica e di impatto sul budget, applicati a diversi scenari, hanno mostrato un potenziale ritorno dell'investimento a sei anni dall'introduzione dello screening, nonostante la previsione di importanti sforzi economici e organizzativi iniziali.
Le interviste, condotte con professionisti sanitari, persone affette da FC, loro familiari e cittadini, hanno rivelato in generale un atteggiamento positivo nei confronti dell'introduzione di uno screening del portatore. Ciononostante, sono emersi importanti richiami a considerare possibili ricadute sociali, in particolare il rischio che le persone con fibrosi cistica possano sentirsi stigmatizzate o escluse dalla società a seguito di una maggiore consapevolezza della frequenza dei portatori.
Considerando la mancanza di informazioni dettagliate su aspetti importanti dello screening - sia dal punto di vista organizzativo che dell'impatto sociale - i ricercatori hanno suggerito che uno studio pilota condotto su un'area territoriale ampia sarebbe fondamentale. Un tale studio permetterebbe di definire l'operatività ottimale di un eventuale programma di screening, tenendo conto delle specificità del contesto italiano e mitigando i potenziali effetti negativi.
I test genetici pre-concepimento e prenatali, come offerti da laboratori specializzati, consentono già oggi di verificare se uno o entrambi i futuri genitori sono portatori della mutazione del gene CFTR. Essere portatore, come già detto, non significa essere malati, ma implica la possibilità di trasmettere la mutazione ai figli. Informarsi, fare prevenzione e affidarsi a professionisti qualificati significa scegliere di affrontare ogni passo di questo percorso con competenza, consapevolezza e serenità, riducendo le incertezze legate alla fibrosi cistica nel neonato.
Il Ruolo Fondamentale delle Associazioni e della Ricerca Collaborativa
La lotta alla fibrosi cistica è un esempio lampante di come la collaborazione tra istituzioni scientifiche, operatori sanitari, pazienti e familiari, spesso organizzati in associazioni di volontariato, sia cruciale per il progresso scientifico e il miglioramento della qualità della vita. Queste entità svolgono un ruolo insostituibile nel sostegno alla ricerca, nella tutela dei diritti dei malati e nella sensibilizzazione dell'opinione pubblica.
La Lega Italiana Fibrosi Cistica (LIFC) è un'associazione di persone con fibrosi cistica e familiari, con sedi diffuse su tutto il territorio nazionale. Da decenni, la LIFC collabora attivamente con i Centri fibrosi cistica per il miglioramento delle cure e dell'assistenza sanitaria, battendosi per la tutela dei diritti e per il miglioramento della qualità della vita delle persone affette. La sua partecipazione al progetto HTA del Mario Negri, coinvolgendo direttamente alcune persone dell'associazione nelle interviste, ha fornito una prospettiva preziosa, dando voce all'esperienza vissuta dai pazienti e dalle loro famiglie. LIFC, insieme a SIFC (Società Italiana Fibrosi Cistica), ha anche patrocinato studi importanti, come quello multicentrico su adolescenti e Fibrosi Cistica, presentato al 37° ECFS Conference a Göteborg.
La Fondazione per la Ricerca sulla Fibrosi Cistica (FFC Onlus) è un'altra entità di primaria importanza. Con sedi regionali e locali e gruppi di volontari - tra cui persone con FC e familiari - si occupa attivamente di raccogliere fondi. Questi fondi sono poi destinati a sostenere progetti di ricerca che mirano a comprendere meglio la malattia e a sviluppare nuove terapie. Il progetto HTA menzionato in precedenza è stato sostenuto proprio grazie a questi fondi raccolti. La FFC Onlus, in sinergia con istituti di ricerca di eccellenza, come l'Istituto G. Gaslini, dimostra un impegno costante nel far progredire la ricerca verso soluzioni innovative.
Pediatria, arriva il test del sudore per la fibrosi cistica
L'impegno congiunto di queste associazioni e degli enti di ricerca ha portato a risultati significativi. La ricerca sulla fibrosi cistica ha imboccato una strada molto promettente che punta alla correzione del difetto di base della proteina CFTR, responsabile dell'insorgere della malattia. Questa prospettiva, che include farmaci "intelligenti" in grado di agire alla radice della malattia attraverso la correzione e il potenziamento della proteina CFTR difettosa - come i modulatori che intervengono sulla mutazione F508, la più frequente - e le future promesse della terapia genica e dell'editing molecolare, è resa possibile anche dal supporto costante e dalla dedizione di queste organizzazioni.
Il loro ruolo non è solo finanziario, ma anche di catalizzatore sociale e etico. Attraverso il loro operato, le associazioni contribuiscono a sensibilizzare l'opinione pubblica sulla fibrosi cistica, a combattere lo stigma e a promuovere politiche sanitarie che garantiscano ai pazienti l'accesso alle migliori cure e una qualità di vita sempre più elevata. La storia di giovani pazienti come Asia, che condividono le proprie testimonianze di convivenza con la patologia, lanciando messaggi di speranza, è un esempio di come l'impegno di queste associazioni si traduca in un impatto reale sulla vita delle persone. L'obiettivo comune è garantire a chi convive con la fibrosi cistica non solo una maggiore sopravvivenza, ma soprattutto una migliore qualità di vita, con la fiducia che la ricerca collaborativa possa portare a una cura definitiva.