L'atresia intestinale fetale, così come la stenosi, rappresenta una condizione medica di significativa importanza nel campo della pediatria e della chirurgia neonatale. Queste patologie congenite, individuabili già durante la gravidanza, possono avere un impatto profondo sulla salute del neonato, rendendo necessaria una diagnosi tempestiva e un intervento chirurgico mirato. Con il termine atresia si indica una malformazione congenita di un organo cavo, contraddistinta dall'occlusione di un canale o di un orifizio normalmente aperti. Questa interruzione nella pervietà può interessare diversi organi canaliformi, come l'esofago e l'intestino, o orifizi organici come l'ano e la vagina. Nel contesto intestinale, atresia significa che un tratto dell'intestino è "chiuso" o addirittura mancante, impedendo la progressione del cibo al suo interno. Similmente, la stenosi implica un restringimento parziale di un tratto intestinale, che ostacola ma non blocca completamente il transito.
Questa complessa condizione è un argomento di grande interesse per genitori, tutori e professionisti sanitari. Comprendere appieno le sue cause, i sintomi che possono manifestarsi sia prima che dopo la nascita, e le opzioni terapeutiche disponibili, è fondamentale per garantire il migliore esito possibile per i bambini affetti. La gestione di queste patologie non si limita all'aspetto medico e chirurgico, ma si estende anche al supporto psicologico e pratico per la famiglia, riconoscendo l'importanza del contatto e della relazione nell'immediato post-partum.
Comprendere l'Atresia: Una Malformazione Congenita
L'atresia, dal greco "a-tresis" che significa "mancata perforazione", descrive una condizione in cui una parte del corpo che dovrebbe essere aperta è invece chiusa o non si è formata correttamente. Nel caso specifico dell'atresia intestinale, un tratto dell'intestino è mancante o non si forma correttamente durante lo sviluppo fetale, provocando un'ostruzione. Questa interruzione congenita del tubo digerente fa sì che il transito si interrompa o non si instauri. La patologia può variare in gravità da caso a caso e può coinvolgere diverse parti dell'intestino, ma le sedi più colpite sono generalmente l'ileo e il digiuno, che sono due tratti dell'intestino tenue. In alcuni casi, l'intestino può essere chiuso da un'area stenotica, ovvero un restringimento, o da una membrana che impedisce il passaggio dei contenuti intestinali. È una malattia rara, che consiste nell'assenza o ostruzione di un tratto di intestino che porta all'occlusione intestinale.
Esistono diverse tipologie di interruzione. Ci può essere la presenza di una membrana che divide l'intestino in due sezioni, fungendo come una strada sbarrata, oppure i monconi possono essere completamente separati o congiunti da passaggi molto ristretti. Esistono anche casi di atresia multipla con più interruzioni, anche di diverso tipo, che rendono la situazione clinica ancora più complessa.
Le Diverse Manifestazioni dell'Atresia
L'atresia può interessare vari organi, non solo l'intestino. La sua classificazione aiuta a comprendere la specificità di ogni condizione e le relative implicazioni cliniche.
Atresia Esofagea: Un'Interruzione delle Vie Digestive Superiori
L'atresia esofagea si caratterizza per una incompleta o alterata formazione dell'esofago. Esistono 5 tipi principali di atresia esofagea, la maggior parte dei quali è caratterizzato dalla presenza di una fistola tra trachea ed esofago. Questa condizione impedisce al cibo di raggiungere lo stomaco, richiedendo un intervento chirurgico immediato dopo la nascita.
Atresia Duodenale: L'Ostruzione della Prima Parte dell'Intestino Tenue
L'atresia duodenale è una patologia congenita che colpisce l'apparato digerente, in particolare il duodeno, la prima parte dell'intestino tenue. È l'ostruzione completa del duodeno nel bambino ed è individuabile già durante i primi mesi di gravidanza. Questa condizione è importante perché può portare a gravi complicazioni se non diagnosticata e trattata tempestivamente. L'atresia duodenale è un difetto congenito caratterizzato dalla formazione incompleta del duodeno, che causa un blocco che impedisce al cibo di passare attraverso il tratto digerente. Questa anomalia rappresenta la seconda atresia più frequente dell'apparato gastrointestinale, con un'incidenza stimata di circa 1 su 10.000 nati vivi. Individuabile già durante la gravidanza, essa interessa 1 neonato su 7000 nati vivi.
L'atresia duodenale è dovuta alla mancata canalizzazione del duodeno embrionario. Questa mancata canalizzazione può essere correlata a un evento ischemico o a fattori genetici. A differenza di altre atresie intestinali, l'atresia duodenale è comunemente associata ad altre anomalie congenite. Circa il 25-40% dei neonati con atresia duodenale presenta la sindrome di Down (Trisomia 21). Altre anomalie associate comprendono l'associazione VACTERL (anomalie vertebrali, atresia anale, malformazioni cardiache, fistola tracheoesofagea, anomalie renali e aplasia radiale, e anomalie degli arti), malrotazione, pancreas anulare, anomalie del tratto biliare e anomalie mandibolo-facciali.
Atresia Intestinale: Interruzione del Digiuno e dell'Ileo
Nell'atresia intestinale, come precedentemente menzionato, un tratto dell'intestino tenue (più frequentemente digiuno o ileo) è mancante o non si forma correttamente. Questa condizione porta all'impossibilità di progressione del cibo al suo interno. A causa dell'atresia, il liquido amniotico che il feto normalmente deglutisce non può progredire completamente attraverso l'intestino. Se ci limitiamo a considerare la forma di atresia intestinale localizzata nel digiuno, la parte preceduta dal duodeno, si può dire che è una malformazione che interessa tra lo 0,5 e 1 bambino ogni 10.000 nati.
Atresia Biliare e Polmonare: Altre Forme Rilevanti
Nell'atresia biliare si verifica la mancata formazione di una parte delle vie biliari oppure il loro progressivo restringimento, fino alla loro occlusione, subito dopo la nascita. Questa condizione impedisce il corretto drenaggio della bile, portando a ittero e danni epatici.
L'atresia polmonare è un difetto cardiaco congenito che interessa la valvola polmonare, deputata all'uscita del sangue dal ventricolo destro verso i polmoni. La valvola polmonare nello specifico, può essere quasi o completamente chiusa. Questa struttura, necessaria durante lo sviluppo del feto nell'utero, si chiude normalmente dopo la nascita. Per questo, se nei primi minuti di vita al neonato non si somministra un farmaco (prostaglandine per via endovenosa) che mantenga il dotto arterioso aperto, le conseguenze possono essere fatali. Il trattamento chirurgico è altresì necessario per l'atresia polmonare, molto spesso più di uno, nei primi anni di vita.
Atresia Anale: Difetti nell'Orifizio Anale
L'atresia anale si caratterizza per la presenza di tessuto che chiude l'ano, e che può essere spesso diversi centimetri o sottile come una membrana. Questa malformazione congenita impedisce l'evacuazione delle feci e richiede un intervento chirurgico correttivo.
Altre Cause di Ostruzione Duodenale
Oltre all'atresia vera e propria, il duodeno può essere ostruito per stenosi o per compressione da una massa estrinseca. Anche queste condizioni sono spesso associate alla sindrome di Down e richiedono un approccio diagnostico e terapeutico simile all'atresia duodenale.
Stenosi Duodenale
La stenosi duodenale è un'anomalia meno frequente dell'atresia duodenale ma si presenta in modo analogo, causando un restringimento parziale del duodeno che ostacola il transito intestinale. Anch'essa è spesso associata alla sindrome di Down e richiede un intervento chirurgico per ripristinare la pervietà completa del tratto.
Pancreas Anulare
Il pancreas anulare è un'anomalia congenita rara, con un'incidenza stimata tra 5 e 15 su 100.000 nati vivi. Questa condizione è spesso associata alla sindrome di Down. In questa anomalia, il tessuto pancreatico circonda la seconda porzione del duodeno, causando un'ostruzione duodenale di vario grado. Circa due terzi delle persone affette rimangono asintomatiche. Tra coloro che sviluppano i sintomi, la maggior parte li presenta nel periodo neonatale, ma la presentazione clinica può essere ritardata fino all'età adulta. I neonati con pancreas anulare presentano problemi di alimentazione e vomito, che può essere biliare.
La diagnosi di pancreas anulare può essere suggerita da una radiografia dell'addome che mostra lo stesso segno della "doppia bolla" visto nell'atresia duodenale. La diagnosi può essere fatta anche con una RX del tratto gastrointestinale superiore e con maggiore precisione con una TC o una colangiopancreatografia in RM. La colangiopancreatografia retrograda endoscopica (CPRE) può essere eseguita in bambini più grandi per una diagnosi più dettagliata. Il trattamento del pancreas anulare è un bypass chirurgico, che può essere realizzato tramite una duodeno-duodenostomia, una duodeno-digiunostomia o una gastro-digiunostomia, aggirando così l'ostruzione causata dal tessuto pancreatico.
Cisti del Coledoco
Una cisti del coledoco può determinare ostruzione duodenale da compressione estrinseca. L'incidenza di questa condizione varia significativamente, da circa 1 su 100.000 persone negli Stati Uniti o in Europa a 1 su 13.000 in Giappone, dove oltre la metà dei casi segnalati si verifica. Ci sono alcune prove che l'incidenza delle cisti del coledoco sia in aumento.
I neonati con cisti del coledoco possono presentare una classica triade di sintomi, sebbene difficili da desumere in un neonato: dolori addominali, una massa palpabile nel quadrante superiore destro e ittero. Se la cisti è ampia, può essere presente anche ostruzione duodenale di grado variabile. I neonati possono manifestare colestasi, e in alcuni lattanti, la pancreatite è un reperto associato.
La cisti del coledoco è diagnosticata tramite ecografia, generalmente. Queste cisti possono essere ulteriormente definite mediante RM colangiopancreatografica, colangiopancreatografia retrograda endoscopica (CPRE) o ecografia endoscopica. Il trattamento della cisti del coledoco è chirurgico e richiede la rimozione completa della cisti a causa dell'alto rischio (7-30%) di sviluppare un cancro nei residui della cisti. La procedura chirurgica più comunemente utilizzata è un'epatico-digiunostomia su ansa ad Y secondo Roux, tipicamente effettuata in laparoscopia. Alcuni studi recenti hanno rilevato che il risultato può essere migliorato con la chirurgia robot-assistita. È importante sottolineare che il rischio di cancro è ridotto ma non completamente eliminato con la resezione completa.
Cause e Fattori di Rischio: Un Quadro Complesso
La causa prima dell'atresia intestinale è ad oggi ancora sconosciuta, sebbene esistano diverse teorie volte a spiegarne l'insorgenza. Tuttavia, si ritiene che una combinazione di fattori genetici e ambientali possa contribuire allo sviluppo di queste malformazioni congenite.
Fattori Genetici e Sindromici
Si ritiene che i fattori genetici contribuiscano in modo significativo allo sviluppo dell'atresia duodenale e di altre atresie intestinali. La condizione è spesso associata ad anomalie cromosomiche, in particolare alla trisomia 21 (sindrome di Down). Come già menzionato, circa il 25-40% dei neonati con atresia duodenale ha la sindrome di Down. Anche altre sindromi genetiche, come l'associazione VACTERL (un gruppo di difetti congeniti che include anomalie vertebrali, atresia anale, malformazioni cardiache, fistola tracheoesofagea, anomalie renali e aplasia radiale, e anomalie degli arti), possono includere l'atresia duodenale come componente. Sebbene l'atresia duodenale non sia direttamente ereditaria nel senso comune del termine, pur ricorrendo con maggiore frequenza in alcune situazioni particolari, fattori genetici e sindromi ad essa associati possono essere ereditari.
In alcuni casi, ci può essere un'associazione tra l'atresia intestinale e la fibrosi cistica, una malattia che a causa di un difetto enzimatico porta alla formazione di secrezioni mucose molto più dense. Questo può contribuire all'occlusione intestinale.
Fattori Ambientali e Stile di Vita Materno
Sebbene la causa esatta dell'atresia duodenale non sia ancora del tutto chiara, alcuni studi suggeriscono che i fattori ambientali durante la gravidanza possano svolgere un ruolo. Ad esempio, infezioni materne o l'esposizione a determinati farmaci o sostanze potrebbero potenzialmente aumentare il rischio di anomalie congenite, tra cui l'atresia duodenale. Alcune scelte di vita e abitudini alimentari durante la gravidanza possono influenzare il rischio di patologie congenite. Ad esempio, un'assistenza prenatale inadeguata, una cattiva alimentazione e l'abuso di sostanze (come il fumo o il consumo di alcol) possono avere un impatto negativo sullo sviluppo fetale, predisponendo a varie malformazioni.
Malformazioni Associate
Oltre alle sindromi genetiche, le atresie intestinali possono coesistere con altre problematiche a carico dell'intestino, quali la malrotazione intestinale o la duplicazione intestinale. La malrotazione intestinale è un'anomalia che si verifica nella fase di incorporazione nell'addome delle strutture embrionali che diventeranno l'intestino. A volte questo processo si complica e l'organo non si fissa come dovrebbe. Questo predispone a una torsione intestinale anomala, chiamato volvolo, in cui l'intestino e i suoi vasi sanguigni sono come se venissero improvvisamente strozzati. Si creano così delle aree necrotiche che dovranno essere rimosse il prima possibile dopo la nascita.
Un'altra causa di occlusione intestinale nel neonato può essere l'enterocolite ulceronecrotizzante del prematuro, un'infiammazione della mucosa intestinale con ulcere e piaghe da necrosi, che colpisce solitamente bambini venuti alla luce prima della trentaduesima settimana di gestazione o sottoposti a una rianimazione difficoltosa alla nascita.
Diagnosi Prenatale e Postnatale: Identificare la Condizione
La diagnosi precoce è cruciale per la gestione efficace dell'atresia e della stenosi intestinale, migliorando significativamente la prognosi del neonato.
Diagnosi Prenatale
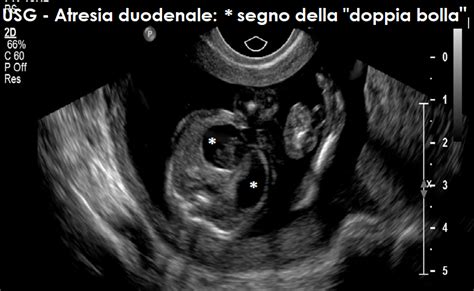
L'atresia intestinale è una condizione diagnosticabile prima della nascita. In epoca prenatale, la diagnosi di atresia duodenale è sospettata in presenza di polidramnios (un eccesso di liquido amniotico) e/o stomaco dilatato. A causa dell'atresia, il liquido amniotico che il feto normalmente deglutisce non può progredire completamente attraverso l'intestino, causando il suo accumulo. Così, in genere, all'esame ecografico fetale del secondo trimestre si evidenzia una dilatazione dello stomaco e delle prime anse intestinali, che tende a peggiorare progressivamente fino al termine della gravidanza. L'ecografia prenatale può rilevare un segno caratteristico noto come "doppia bolla" (una grande bolla gastrica e una più piccola bolla duodenale prossimale) fino nell'80% dei casi. Il polidramnios tende talvolta ad accelerare i tempi del parto. È importante sapere che il feto non risente di tale situazione finché si trova nella pancia della mamma, poiché è la madre ad assicurargli il nutrimento.
Diagnosi Postnatale
Nel periodo postnatale, i neonati con atresia duodenale presentano tipicamente sintomi come difficoltà di alimentazione e vomito, che può essere biliare. Anche il ritardo o l'assenza dell'evacuazione di meconio si osserva spesso in presenza di atresia del tenue, come anche nella malattia di Hirschsprung, nella fibrosi cistica e quando il colon è di ridotte dimensioni. L'occlusione intestinale, spesso dolorosa, può scatenare il pianto nel neonato.
La diagnosi inizia in genere con una valutazione clinica approfondita del bambino. La diagnosi è suggerita dai sintomi e dal riscontro RX della classica 'doppia bolla' visibile in una radiografia addominale: una bolla nello stomaco e l'altra nel duodeno prossimale. Nell'intestino distale, l'aria è scarsa o assente, indicando l'ostruzione.
Per l'atresia intestinale generale, la diagnosi viene eseguita attraverso una radiografia addominale, per individuare le anse dell'intestino tenue dilatate e rilevare l'ostruzione. L'esame clinico del bambino è fondamentale. Talvolta l'esplorazione rettale si rivela utile per mettere in evidenza una stenosi o per provocare un'emissione di meconio, soprattutto nella malattia di Hirschsprung.
Sebbene una serie di indagini radiologiche sul tratto gastrointestinale superiore possa fornire una diagnosi definitiva, essa deve essere effettuata con attenzione da un radiologo esperto che esegua questa procedura sui bambini, al fine di evitare l'aspirazione. Generalmente non è necessaria se l'intervento chirurgico sta per essere eseguito immediatamente. Se la chirurgia deve essere ritardata (ad esempio, a causa di altri problemi medici, come la sindrome da distress respiratorio o la necessità di stabilizzare il lattante), un clisma opaco deve essere eseguito per confermare che il segno della doppia bolla non è dovuto a malrotazione. La conferma della diagnosi avviene in un contesto chirurgico specializzato, dove si effettuano esami radiologici complementari (per lo più radiografia semplice dell'addome). Per la diagnosi della malattia di Hirschsprung è necessaria una biopsia rettale.
I sintomi della perforazione intestinale postnatale, una grave complicazione, compaiono solitamente molto presto, spesso entro le prime 24-48 ore di vita, sebbene in caso di stenosi (restringimento parziale) possano manifestarsi più tardi. Questi includono un gonfiore addominale, dove l'addome appare teso, lucido e marcatamente dilatato. Esami del sangue sono necessari per valutare l'entità dell'infiammazione e dello squilibrio metabolico. Nella maggior parte dei casi, la diagnosi avviene mentre il neonato è ancora in ospedale dopo la nascita.
Trattamento Chirurgico: La Soluzione Definitiva
Il trattamento primario per l'atresia e la stenosi intestinale è l'intervento chirurgico. La chirurgia è la terapia definitiva per risolvere l'ostruzione e ripristinare la continuità del tratto digerente.
Preparazione all'Intervento
Una volta sospettata la patologia, il neonato non deve più assumere nulla per bocca. Per aiutare il bambino a svuotare lo stomaco e l'intestino prevenendo il vomito, si deve posizionare un sondino naso-gastrico per decomprimere lo stomaco. Per nutrirlo e assicurargli un adeguato apporto di liquidi, si utilizza la via endovenosa (fleboclisi). Spesso il trattamento medico viene posto in atto immediatamente, includendo anche l'antibioticoterapia in alcuni casi. L'atresia duodenale di per sé non richiede l'espletamento di un parto cesareo; è auspicabile che il bambino possa nascere a termine di gravidanza, ove possibile, da parto spontaneo. L'atresia duodenale può essere corretta chirurgicamente alla nascita.
Non essendo l'atresia intestinale una vera emergenza acuta, si attenderà che il bambino raggiunga un buon equilibrio prima di procedere all'intervento chirurgico. Gli approcci terapeutici possono variare in base all'età e allo stato di salute generale del paziente.
La Procedura Chirurgica
L'operazione consiste nell'eliminare il tratto chiuso dell'intestino interessato e nel ripristinare la continuità. Nella gran parte dei casi, si riesce a risolvere il problema con un unico intervento. Con la chirurgia si eliminano eventuali membrane di separazione e si collegano i diversi segmenti di intestino. Si utilizzano tecniche di congiungimento particolari perché spesso i differenti monconi hanno calibri diversi. Le suture vanno eseguite con meticolosità, altrimenti si aumenta il rischio di incorrere in possibili fistole.
L'accesso per l'intervento solitamente è l'ombelico, in cui si inserisce un laparoscopio per avere una visuale interna. Dall'apertura si espongono le anse intestinali e ci si concentra sulle aree interessate. Le tecniche chirurgiche si stanno evolvendo, e oggi, grazie a procedure molto avanzate, si può ottenere una vita normale anche nei casi in cui il tratto di intestino sano sia molto breve. Il trattamento chirurgico è indicato in presenza di lesioni estese: correzione della torsione dell'intestino tenue, ablazione del segmento leso e abboccamento delle parti sane del tubo digerente e così via. L'obiettivo della chirurgia è riparare la perforazione e risolvere l'ostruzione (atresia o stenosi) tramite resezione e anastomosi: rimozione del segmento intestinale atresico e della zona perforata, seguita dall'unione (anastomosi) dei due monconi sani.
Gestione Post-Operatoria e Recupero
Dopo l'intervento chirurgico, il periodo di recupero è cruciale e richiede cure attente e personalizzate, spesso in un ambiente di terapia intensiva.
Cura del Neonato e Nutrizione
Il neonato viene assistito in terapia intensiva. La nutrizione parenterale totale (NPT) è essenziale finché l'intestino non riprende la sua normale funzione (canalizzazione). Questo significa che il bambino riceve tutti i nutrienti necessari direttamente in vena attraverso un catetere venoso centrale. Dopo l'intervento chirurgico, i neonati potrebbero necessitare di formule specifiche o di una graduale reintroduzione degli alimenti.
In genere, una volta ripreso il transito intestinale (il numero di giorni è variabile), la mamma comincerà gradualmente ad alimentare il bambino seguendolo nei piccoli passi che lo porteranno lentamente ad assumere una quantità di latte sufficiente alla sua crescita. Qualora le atresie fossero multiple o l'intestino residuo fosse molto breve, i tempi di recupero possono allungarsi notevolmente, e può essere necessaria l'alimentazione artificiale parenterale (attraverso un catetere venoso centrale), enterale (attraverso il sondino naso-gastrico) o mista.
La ripresa può apparire lenta, ma il bambino ha i suoi ritmi e bisogna imparare a seguirli e a riconoscerli. Il corpo ha i suoi ritmi e noi dobbiamo solo imparare a riconoscerli e a seguirli. Le complicazioni a breve termine possono includere infezioni del sito chirurgico e ritardi nella guarigione.
Il Ruolo Fondamentale dei Genitori: Supporto e Cura
Le cure di cui il neonato ha bisogno limitano e modificano il naturale contatto madre-padre-neonato nell'immediato post-partum. Tuttavia, è di vitale importanza mantenere e rafforzare questo legame. Il reparto è aperto ai genitori 24 ore su 24, a eccezione di alcuni momenti particolari. È importante sapere che il reparto è aperto ai genitori 24 ore su 24, fatti salvi alcuni momenti particolari. Possiamo dire che esiste un bisogno reciproco di vicinanza e di relazione. Sia prima che dopo l'intervento è importante la presenza dei genitori: il bambino li riconosce e ha bisogno di essere in contatto con loro.
I genitori avranno quindi un po' di tempo per stare accanto a lui: in queste prime ore è il papà che garantisce al bambino un primo contatto, potendo stare accanto al bambino nelle ore che precedono l'intervento chirurgico. È bene sapere che il bambino appena nato può riconoscere la voce del papà e sentirne la presenza.
È bene che i genitori inizino presto ad accudire il bambino (cambio pannolino, tutina ecc): nessuno lo può fare con più amore e attenzione. Se la presenza della flebo e del catetere venoso o vescicale creano disagio, le infermiere mostreranno come è meglio prendere il neonato in braccio. I genitori, prima del parto, possono anche visitare il reparto dove sarà accolto e curato il bambino alla nascita in modo da familiarizzare con le persone e l'ambiente, contribuendo a ridurre l'ansia e a prepararsi meglio all'esperienza.

Complicazioni: Perforazione Intestinale Postnatale
La perforazione intestinale postnatale dovuta ad atresia o stenosi intestinale è una condizione chirurgica d'emergenza estremamente grave che colpisce i neonati nei primi giorni o settimane di vita. Quando il contenuto intestinale (gas, secrezioni digestive e meconio) non può progredire a causa del blocco, le pareti dell'intestino si dilatano eccessivamente. Questa dilatazione compromette l'apporto di sangue alla parete (ischemia), rendendola fragile fino a causarne la perforazione. Questa condizione richiede un intervento tempestivo poiché la perforazione espone il neonato a rischi sistemici immediati, tra cui la sepsi neonatale e lo shock ipovolemico. Le cause alla base della perforazione intestinale in presenza di atresia o stenosi sono strettamente legate allo sviluppo embrionale del feto.
I sintomi, come il gonfiore addominale, compaiono solitamente molto presto, spesso entro le prime 24-48 ore di vita. Il trattamento, come descritto in precedenza, include la resezione e l'anastomosi del tratto intestinale compromesso, con assistenza post-operatoria in terapia intensiva e nutrizione parenterale totale.
Prognosi e Prospettive Future
Con una diagnosi precoce e un intervento chirurgico appropriato, la prognosi per i neonati con atresia duodenale è generalmente favorevole. Anche per le atresie intestinali in generale, la prognosi di questi interventi è generalmente buona. È importante notare che la prognosi è generalmente buona se l'intervento avviene tempestivamente e se non sono presenti altre malformazioni gravi, specialmente quelle cardiache, che possono complicare il quadro clinico.
Non esiste una prevenzione specifica per l'atresia o la stenosi intestinale, trattandosi di anomalie congenite dello sviluppo. L'atresia duodenale è una grave condizione congenita che richiede una diagnosi e un trattamento tempestivi. Comprendere le sue cause, i sintomi e le opzioni terapeutiche può aiutare genitori e tutori a richiedere tempestivamente assistenza medica, garantendo ai neonati le migliori possibilità di recupero e una vita sana.
Disclaimer: Questo articolo ha solo scopo informativo e non sostituisce il parere di un medico.
tags: #stenosi #intestinale #fetale