Le neurofibromatosi rappresentano un gruppo eterogeneo di malattie genetiche caratterizzate da una predisposizione significativa allo sviluppo di tumori che interessano il sistema nervoso, sia centrale che periferico. Queste condizioni, sebbene rare individualmente, rivestono un'importanza cruciale per l'impatto profondo che possono avere sulla vita degli individui affetti e delle loro famiglie. Comprendere la loro natura genetica, le modalità di trasmissione e le opzioni diagnostiche, inclusa la diagnosi prenatale, è fondamentale per una gestione efficace e per la pianificazione familiare. L'evoluzione della genetica molecolare ha reso disponibili strumenti diagnostici avanzati, che permettono una identificazione accurata delle mutazioni responsabili, guidando così decisioni cliniche e fornendo supporto essenziale.
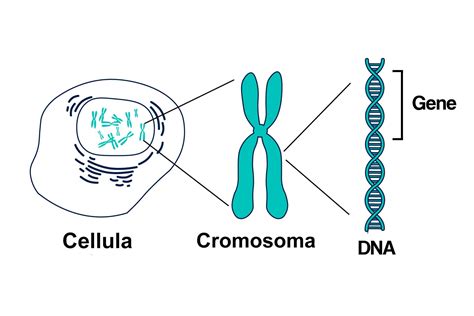
Il Fondamento Genetico: Cromosomi, Geni e Mutazioni
Per comprendere appieno le neurofibromatosi e la loro ereditarietà, è essenziale richiamare alcuni concetti fondamentali della genetica umana. Il nostro corpo è costituito da milioni di cellule; ognuna di esse contiene delle strutture chimiche chiamate cromosomi. Nell'uomo, in ogni cellula somatica, ci sono 46 cromosomi raggruppati in 23 paia. Di queste, 22 paia sono autosomi, ovvero cromosomi che non determinano il sesso, e un paio sono cromosomi sessuali (XX per le femmine e XY per i maschi). I geni, componenti essenziali del nostro patrimonio ereditario, sono piccole parti del cromosoma, una specie di maglia cromosomica. Anche i geni vanno a due a due; ci sono circa 35000 geni distribuiti lungo i 46 cromosomi, in un ordine molto specifico e preciso.
I geni dirigono il comportamento della cellula, agendo come istruzioni per la produzione di proteine e per il controllo dei processi cellulari. Quando un gene è attivato, una varietà di eventi può prodursi nella cellula, a seconda della funzione particolare di questo gene. Qualche gene è responsabile di tratti evidenti quali il colore degli occhi; altri controllano la produzione di sostanze essenziali alle reazioni chimiche del nostro corpo, o regolano la crescita e lo sviluppo cellulare. È importante notare che alcune parti del cromosoma non sono geni, ma soltanto “simboli di punteggiatura” tra geni, con funzioni regolatorie o strutturali non direttamente codificanti.
Una mutazione è un cambiamento nella sequenza del DNA di un gene. Mutazioni di geni si producono da sempre e continuano a prodursi. Questo processo è una parte naturale dell'evoluzione biologica, e la maggior parte delle mutazioni non sono scopribili e alcune non nuocciono. Tuttavia, alcune mutazioni possono alterare significativamente la funzione di un gene, portando allo sviluppo di malattie genetiche. Nel contesto delle neurofibromatosi, queste mutazioni interessano geni specifici che codificano per proteine regolatrici della crescita cellulare, con conseguenze patologiche. La comprensione di queste alterazioni a livello molecolare è la chiave per la diagnosi e per la ricerca di terapie mirate.
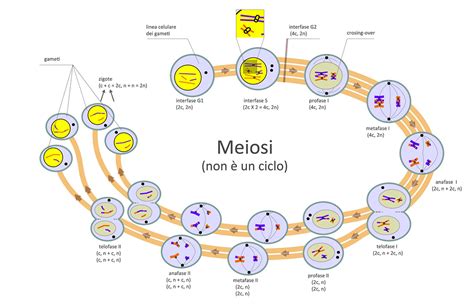
Il Processo di Meiosi e la Trasmissione Ereditaria
La trasmissione delle caratteristiche genetiche da una generazione all'altra avviene attraverso un processo specializzato di divisione cellulare chiamato meiosi. Le cellule germinali (ovuli nelle femmine e spermatozoi nei maschi), prima di raggiungere la maturità, contengono 23 paia di cromosomi, ossia il patrimonio genetico di ogni altra cellula del corpo. Quando raggiungono la maturità, queste cellule subiscono un processo speciale, chiamato meiosi: il risultato è che ogni cellula matura (ovulo come spermatozoo) possiede ora solo la metà del patrimonio genetico iniziale, cioè 23 cromosomi singoli. Questo dimezzamento del numero di cromosomi è fondamentale per mantenere costante il numero di cromosomi della specie dopo la fecondazione, quando lo spermatozoo e l'ovulo si uniscono, ripristinando il corredo di 46 cromosomi (23 paia) nel nuovo individuo.
Questo meccanismo è cruciale per la trasmissione autosomica dominante, che è la modalità di trasmissione delle neurofibromatosi di tipo 1 (NF1) e tipo 2 (NF2). Una malattia con trasmissione autosomica dominante significa che è provocata da un solo gene “dominante” che ha subito una mutazione. Se un genitore possiede un allele mutato dominante per una data condizione, vi è un rischio del 50% ad ogni gravidanza che il gene mutato venga trasmesso ai figli. Questo gene mutato può: sia essere trasmesso da un genitore che ha lui stesso la NF1, sia apparire per caso in un individuo nella cui famiglia nessuno ha la NF1, tramite una mutazione spontanea (o "de novo"). Quando un individuo possiede il gene mutato della NF1, sia per eredità che per “mutazione spontanea”, c’è una possibilità su due che egli lo trasmetta ad ognuno dei suoi bambini. C’è ovviamente, pertanto, anche una possibilità su due che il gene non venga trasmesso; è una sorta di testa o croce.
La Neurofibromatosi di Tipo 1 (NF1): Una Malattia Multisistemica
La neurofibromatosi di tipo 1 (NF1), precedentemente nota come malattia di Von Recklinghausen, è una malattia genetica multisistemica che colpisce indistintamente uomini e donne di ogni gruppo etnico. È considerata una delle più frequenti malattie rare e colpisce circa 1 su 3500 individui. La modalità di trasmissione è autosomico dominante. Il gene responsabile della NF1 è localizzato sul braccio lungo del cromosoma 17, e le mutazioni in questo gene NF1 sono la causa principale della condizione. Circa il 50% dei pazienti NF1 sono casi sporadici che hanno genitori completamente sani e senza alcuna alterazione nel gene NF1, il che significa che la mutazione è insorta de novo nel paziente.
La NF1 si manifesta con un'ampia gamma di sintomi clinici, che possono variare considerevolmente da persona a persona, persino all'interno della stessa famiglia. Lo stesso gene mutato presente in diversi membri della stessa famiglia (fratello, sorella, genitori, nonni…) può provocare a seconda dei casi dei sintomi molto diversi e di una gravità molto variabile. Così, un genitore affetto da NF1 leggera (poche Macchie Caffelatte o neurofibromi) può avere un bambino molto più affetto da NF1, anche la situazione opposta può esistere. Questa variabilità, nota come espressività variabile, rende la prognosi e la gestione della malattia particolarmente complesse.
Clinicamente, pur essendo una malattia multisistemica, la NF1 colpisce principalmente la cute, con neurofibromi multipli e le tipiche macchie caffelatte. I noduli di Lisch, proliferazioni benigne dell’iride, da ricercarsi all’esame oculistico mediante lampada a fessura, sono un segno più tardivo (>60% dei pazienti). I neurofibromi, che compaiono di solito nella seconda decade di vita, il più delle volte sotto forma di noduli cutanei e sottocutanei (> 50% dei casi), sono un'altra caratteristica distintiva. Oltre a queste manifestazioni cutanee e oculari, i pazienti con NF1 possono presentare problemi oculari, come il glioma delle vie ottiche, e anomalie ossee, come la pseudoartrosi tibiale o la displasia dello sfenoide.
La NF1 è associata anche a disturbi dell’apprendimento e un lieve ritardo cognitivo, convulsioni e ipertensione arteriosa. Un aspetto cruciale della NF1 è l'aumentato rischio di sviluppare sia tumori benigni che maligni. Questi pazienti hanno il 10 per cento di rischio di sviluppare particolari tumori come il già citato glioma delle vie ottiche e tumori maligni della guaina dei nervi periferici (MPNST), ma anche un aumento del rischio di insorgenza di tumore della mammella, feocromocitoma, rabdomiosarcoma e leucemia. Nonostante la complessità e la variabilità della condizione, l’aspettativa di vita è pressoché uguale a quella della popolazione generale, anche se è richiesta una sorveglianza medica costante. I bambini con NF1 hanno bisogno di essere sorvegliati regolarmente (anche se apparentemente sono in buona salute). Due esami annuali sono consigliati durante l’infanzia, realizzati preferibilmente da un medico che conosca bene i problemi della NF1.
La Neurofibromatosi di Tipo 2 (NF2): Un Quadro Clinico Distinto
Completamente diverso è il quadro della neurofibromatosi di tipo 2 (NF2). Questa forma, molto diversa geneticamente dalla NF1, ha anche lei delle caratteristiche cliniche molto particolari; è comunque rara (1 caso ogni 40.000). A differenza della NF1, i segni cutanei (neurofibromi e Macchie Caffelatte) sono il più delle volte discreti e talvolta persino assenti. I noduli di Lisch non si ritrovano nella NF2, rappresentando un chiaro elemento distintivo diagnostico.
La caratteristica principale della NF2 è lo schwannoma al nervo acustico, bilaterale nel 90-95% dei casi, che porta a sordità progressiva, vertigini, acufeni e disturbi dell’equilibrio. Questa condizione predispone anche ad altri tumori del sistema nervoso centrale e periferico, come schwannomi, meningiomi ed ependimomi, che possono insorgere in varie localizzazioni craniche e spinali. In molti casi, la NF2 è anche associata a cataratta giovanile, che può manifestarsi precocemente e influenzare la vista.
La NF2 non presenta alcuna delle altre manifestazioni cliniche della NF1, ma per contro presenta un rischio di sviluppare tumori praticamente del 100 per cento, anche in giovane età. La morbilità è alta, a causa di sordità, paresi facciale, disturbi oculari fino alla cecità, deficit neurologici sede specifici e l’aspettativa di vita è fortemente ridotta nei casi più gravi. Questa marcata differenza clinica, genetica e prognostica ha portato nel 1987 la National Institutes of Health Consensus Conference americana a scindere definitivamente le due condizioni. È quindi errato e fuorviante usare lo stesso termine “neurofibromatosi” per queste due malattie. Purtroppo nel mondo medico è rimasta invece l’idea che siano malattie simili: di fatto, anche secondo il Decreto del Ministro della Salute 279/2001, NF1 e NF2 hanno lo stesso codice di esenzione malattia rara (RBG010), il che può contribuire a questa percezione errata.
La trasmissione della NF2 si fa sul modo autosomico dominante, il che significa che il rischio ad ogni gravidanza di trasmettere il gene è anch'esso di uno su due (50%). Sappiamo che l’anomalia genetica si trova sul cromosoma 22, e il gene della NF2 è stato individuato nel 1993, permettendo una diagnosi molecolare precisa.
Schwannomatosi: La Terza Forma di Neurofibromatosi
Accanto alle neurofibromatosi di tipo 1 e tipo 2, esistono altre forme, dai contorni ancora poco chiari (trattasi di varianti oppure di forme geneticamente distinte?). Una di queste è la neurofibromatosi di tipo 3, oggi chiamata schwannomatosi, che presenta un quadro clinico molto simile alla NF2. Si caratterizza per la presenza di schwannomi multipli e, più raramente, meningiomi, ma tipicamente mai ependimomi, che sono invece più comuni nella NF2 classica.
Inizialmente, una delle caratteristiche principali considerate per la diagnosi della schwannomatosi era l'assenza di schwannoma al nervo acustico. Tuttavia, questa distinzione non è più valida dopo la descrizione di alcuni casi clinici con coinvolgimento del nervo acustico, sebbene questo non si manifesti mai in forma bilaterale, che è invece tipica della NF2. Questa osservazione sottolinea la complessità e la sovrapposizione fenotipica tra le diverse forme di neurofibromatosi.
A livello genetico, nella schwannomatosi il gene NF2 è coinvolto in modo secondario, mentre la mutazione primaria è a carico di un altro gene da esso poco distante, sempre sul braccio lungo del cromosoma 22. I geni più frequentemente implicati sono LZTR1 o SMARCB1. Questa scoperta genetica ha contribuito a distinguere ulteriormente la schwannomatosi come entità separata, pur mantenendo alcune somiglianze cliniche con la NF2, evidenziando l'importanza dell'analisi molecolare per una diagnosi accurata. Esiste anche la neurofibromatosi segmentale, una neurofibromatosi eccezionale dove c’è presenza di neurofibroma e/o Macchie Caffelatte limitate a specifiche regioni corporee.
L'Importanza Cruciale del Test Genetico e della Diagnosi
La diagnosi delle neurofibromatosi si basa essenzialmente sulla storia clinica del paziente e sull'esame obiettivo, data la variabilità e la progressività delle manifestazioni. La familiarità si osserva solo in metà dei casi nella NF1 e NF2 e in non più del 15-25% dei casi di schwannomatosi, il che evidenzia l'alta incidenza di mutazioni de novo e rende la diagnosi genetica ancora più importante in assenza di una storia familiare chiara.
Il test genetico per la Neurofibromatosi (NF1 e NF2) è un’indagine fondamentale per la diagnosi precoce, la gestione familiare e la prevenzione oncologica. Presso il Laboratorio di analisi cliniche CUSMAI, è disponibile il test genetico per la ricerca di mutazioni nei geni NF1 e NF2, eseguito in collaborazione con SYNLAB, il più grande centro di diagnostica al mondo. Il sequenziamento NGS (Next-Generation Sequencing) permette di analizzare in modo completo ed estremamente accurato i geni NF1 e NF2, rilevando mutazioni puntiformi, delezioni, duplicazioni o varianti rare. Questa tecnologia avanzata è in grado di identificare la maggior parte delle mutazioni responsabili, fornendo una diagnosi molecolare precisa. La ricerca di mutazioni nei geni NF1 e NF2 è disponibile presso il Laboratorio CUSMAI, centro di riferimento a Bari e in Puglia per la genetica molecolare clinica.
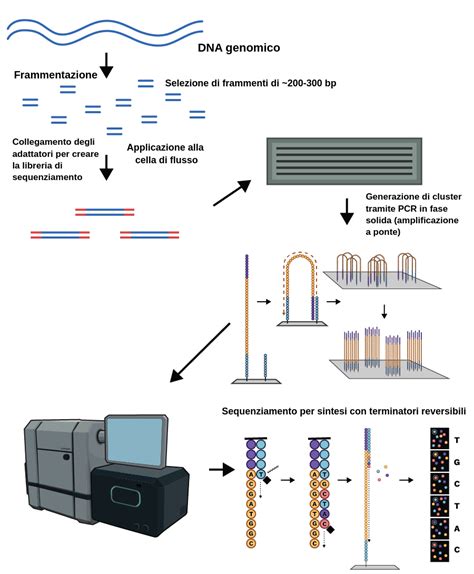
L’analisi genetica si effettua tipicamente sul sangue. In caso di esito negativo su campione ematico, ma con forte sospetto clinico, l'analisi può essere estesa a due diversi tessuti tumorali asportati chirurgicamente, specialmente in presenza di mosaicismo somatico, dove la mutazione non è presente in tutte le cellule del corpo. L'identificazione della mutazione genetica specifica non solo conferma la diagnosi, ma è cruciale per il consiglio genetico, per la stratificazione del rischio e per orientare la sorveglianza clinica.
Diagnosi Prenatale: Opzioni e Considerazioni Etiche
Quando un uomo o una donna, in una coppia, ha la NF, la coppia ha talvolta parecchie difficoltà a decidere se avere o meno un bambino. La disponibilità della diagnosi prenatale offre ai genitori a rischio la possibilità di conoscere lo stato di salute genetica del feto. Nelle famiglie a rischio con mutazione genetica identificata, è possibile effettuare la diagnosi prenatale, attraverso amniocentesi o villocentesi. Queste procedure permettono di prelevare cellule fetali contenenti il DNA del nascituro per l'analisi genetica.
Quando uno dei genitori ha la NF1, c’è un rischio su due ad ogni gravidanza di avere un bambino anch’esso colpito da NF1. Il rischio di avere un bambino che avrà un grave problema di salute può essere stimato al 10% (20% se si considerano solo i bambini con NF). L'importanza dell’afflizione di un soggetto con NF1 varia molto da una persona all’altra, anche all’interno di una stessa famiglia, rendendo le decisioni ancora più complesse. Attualmente la diagnosi prenatale, senza nessuna informazione sull’importanza di una forma eventualmente trasmessa, può essere realizzata facilmente solo quando ci sono varie persone di una stessa famiglia che hanno la NF1 (almeno due persone). Questo sottolinea l'importanza di un'accurata storia familiare e di una consulenza genetica approfondita.
Per calcolare esattamente i rischi di trasmissione, i genitori (apparentemente non colpiti) di un bambino affetto devono certamente farsi esaminare da un medico competente (attento esame della pelle e degli occhi che comprenda la ricerca dei noduli di LISCH) per essere certi che nell’uno nell’altro abbiano la NF1 sotto forma molto attenuata. Se al termine di questo esame, sono riconosciuti immuni da NF1, il rischio di avere un bambino con NF1 scende ad 1 su 3000, un dato che evidenzia il ruolo delle mutazioni de novo nei casi sporadici.
Il consiglio genetico è molto importante: la trasmissione si fa sul modo autosomico dominante (il rischio ad ogni gravidanza di trasmettere il gene è anch’esso di uno su due). I consigli genetici possono aiutare le coppie a prendere la loro decisione; questi consigli genetici non dicono ovviamente alla coppia cosa essa debba fare; si limitano a dare informazioni, a chiarire le situazioni; possono anche spiegare possibilità alternative, ad esempio l’adozione o l’inseminazione artificiale con donatore. Nel caso della NF2 “a mosaico”, dove la mutazione genetica non è presente in tutti i tessuti corporei del genitore, il rischio di trasmissione è più basso del 50%, un'altra variabile da considerare nella consulenza genetica.
Malattie genetiche, clinica, ricerca e uno sportello per le malattie rare
L'incidenza psicologica di questa differenza può essere importante, sia per il bambino affetto, sia per i genitori, rendendo il supporto psicologico e le risorse informative di vitale importanza.
Gestione e Trattamento delle Neurofibromatosi
Attualmente, non esiste una terapia risolutiva per le neurofibromatosi, ma la gestione si concentra sul trattamento sintomatico e sul monitoraggio delle singole problematiche. L'approccio è multidisciplinare e mira a migliorare la qualità di vita dei pazienti e a prevenire o gestire le complicanze.
Nella NF1, il trattamento è spesso chirurgico, per la correzione delle malformazioni ossee e l'asportazione dei neurofibromi cutanei, soprattutto se causano dolore, prurito o problemi estetici. È inoltre fondamentale il supporto psicologico e riabilitativo per i disturbi dell’apprendimento e le disabilità intellettive, che possono avere un impatto significativo sullo sviluppo del bambino. In caso di tumori maligni, come i MPNST, alla chirurgia possono seguire radioterapia e chemioterapia. I costi delle cure per un bambino affetto da NF1 possono essere alti, il che rende essenziale l'accesso a sistemi di supporto e copertura sanitaria adeguati. La NF1 è variabile, imprevedibile ed evolutiva, richiedendo un follow-up medico costante e adattabile alle esigenze individuali del paziente.

Nella NF2 e nella schwannomatosi, il follow-up è basato sulla risonanza magnetica con mezzo di contrasto effettuata periodicamente. Questo monitoraggio è cruciale per identificare precocemente la crescita dei tumori, in particolare gli schwannomi e i meningiomi. L’intervento chirurgico dei tumori cerebrali e midollari è molto rischioso, a causa della loro localizzazione in aree sensibili del sistema nervoso. Per questo motivo, si decide per l’asportazione solo in caso di crescita eccessiva o di importanti deficit neurologici in atto che compromettono la funzione o la qualità di vita del paziente. È importante notare che nei bambini e adolescenti non è indicata la radioterapia per il rischio di induzione di nuovi tumori, il che limita le opzioni terapeutiche in questa fascia d'età. Tuttavia, il farmaco bevacizumab per via endovenosa è usato per cercare di frenare lo sviluppo di schwannomi in rapida crescita, offrendo una speranza per la stabilizzazione della malattia in alcuni pazienti.
Altre Malformazioni Congenite: I Difetti del Tubo Neurale
Oltre alle neurofibromatosi, esistono altre malformazioni congenite che possono essere identificate tramite screening prenatale, sebbene con meccanismi e cause distinte. Sotto l’espressione “difetti del tubo neurale” rientra una vasta gamma di malformazioni congenite. Tutte hanno in comune la comparsa precoce, entro i 28 giorni dal concepimento, un periodo cruciale per lo sviluppo del sistema nervoso. In Italia, i difetti del tubo neurale colpiscono circa 1 bambino ogni 1500 nuovi nati.
I difetti del tubo neurale possono essere “aperti” o “chiusi”, a seconda che ci sia un deficit o un eccesso di tessuti che non si chiudono correttamente. I secondi sono più difficili da individuare e spesso anche meno gravi. La forma più comune di difetto del tubo neurale è la spina bifida. Si tratta di un difetto “aperto”, nel quale il tessuto nervoso di midollo spinale e meningi rimane scoperto.
Le cause dei difetti del tubo neurale sono sia ambientali sia genetiche. Il fattore di rischio più conosciuto e anche più facile da prevenire è la carenza di acido folico durante la gravidanza. Contribuiscono anche l’assunzione di alcuni farmaci antiepilettici, il diabete materno, l’anemia e l’esposizione ad alcune sostanze tossiche.
I comuni test di screening prenatale sono in grado di individuare alcuni difetti del tubo neurale. Ad esempio, l’ecografia può individuare la spina bifida già alla 14a settimana di gestazione, consentendo una diagnosi precoce. Per i difetti chiusi, invece, spesso vengono individuati dopo la nascita e talvolta anche dopo mesi o anni, a causa della loro natura meno evidente. Per il momento non esistono terapie risolutive per i difetti del tubo neurale. Tuttavia, è in via di sviluppo una tecnica per correggere la spina bifida in utero, ma è ancora in fase di sperimentazione, offrendo prospettive future per un intervento precoce e potenzialmente migliorativo.
tags: #neurofibromatosi #test #dna #fetale