Le malformazioni congenite rappresentano una delle principali sfide in ambito pediatrico e ostetrico, interessando una quota significativa della popolazione neonatale. Si stima che la frequenza complessiva dei difetti congeniti si aggiri intorno al 5-6% dei nati, e che queste condizioni interessano circa 8 milioni di bambini nel mondo ogni anno, di cui oltre 25.000 in Italia. Durante le fasi di sviluppo fetale può verificarsi l’insorgenza di malformazioni congenite anche definite anomalie congenite, le quali possono presentarsi con varie manifestazioni cliniche e gravità. Tra queste, le malformazioni craniche fetali rivestono un ruolo di particolare importanza per le loro potenziali implicazioni sulla salute e lo sviluppo neurologico del bambino, e in casi estremi, possono essere associate a esiti infausti come la natimortalità. Comprendere le diverse tipologie, le cause sottostanti e i percorsi diagnostici e terapeutici è essenziale per una gestione efficace di queste condizioni. Questo articolo esplora in dettaglio le malformazioni craniche, i meccanismi che le generano, il fenomeno della natimortalità e le sue eziologie, cercando di delineare le interconnessioni tra questi complessi aspetti della medicina perinatale.
Comprendere le Malformazioni Craniche: Definizione e Caratteristiche Generali
Le malformazioni craniche nei bambini, come la plagiocefalia, la brachicefalia o la scafocefalia, sono una deformazione del cranio in crescita causata dalla pressione meccanica di alcune sue parti prima, durante o dopo il parto. Questa vulnerabilità deriva dalla particolare struttura del cranio fetale e neonatale. Le ossa che formano la volta e la base cranica del feto, del bambino e del neonato di età inferiore ai 3 mesi sono ancora scarsamente ossificate e sono separate l’una dall’altra da suture e fontanelle. Questa caratteristica permette al cranio di adattarsi durante il parto e di espandersi rapidamente per accogliere la crescita cerebrale post-natale. Tuttavia, se per qualsiasi motivo ricevono una pressione eccessiva o continua, ne viene ostacolata la normale crescita e le deformazioni possono essere causate dall’appiattimento di una parte del cranio.
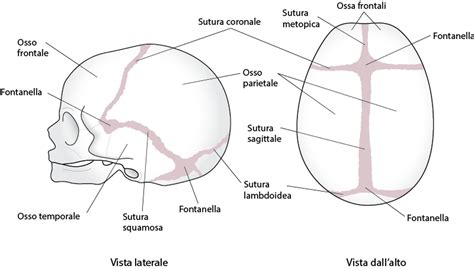
Le malformazioni craniche possono essere suddivise in due grandi categorie: quelle di natura posizionale e quelle derivanti dalla chiusura prematura delle suture craniche, note come craniostenosi. Mentre le prime sono spesso meno gravi e tendono a risolversi con interventi conservativi, le craniostenosi richiedono un approccio più complesso e possono avere conseguenze significative sullo sviluppo neurologico e sulla vita del bambino. L'indice cranico, che valuta il rapporto larghezza-lunghezza del cranio, è un parametro importante per la valutazione di queste deformità: una testa normale è di forma ovale se vista dall’alto e ha un normale rapporto larghezza-lunghezza dell’80%.
La Craniostenosi: Una Complessa Anomalia dello Sviluppo Cranico
Il termine Craniostenosi o Craniosinostosi deriva dal greco e significa chiusura, ristrettezza del cranio, e descrive una condizione caratterizzata dalla prematura saldatura di una o più suture craniche. Questa è una delle malformazioni congenite più frequenti, per la quale viene riportata un’incidenza di circa 1 neonato su 2000. Il cranio è infatti composto da ossa piatte, separate da suture, che si chiudono normalmente fra il primo ed il terzo anno di vita. Man mano che il cervello dei bambini si sviluppa nei primi due anni di vita, raddoppiando il suo volume, le suture permettono al cranio di espandersi progressivamente e grazie a questa “spinta modellante” si ottiene la crescita armonica della testa.
Quando una o più suture sono chiuse prematuramente, il cranio non può espandersi in modo uniforme. Invece, il cranio si espande lungo le suture che rimangono aperte, con una conseguente progressiva deformazione della sua struttura, che comporta una conformazione disarmonica ed anomala del cranio. Questa crescita disarmonica è una caratteristica distintiva della craniostenosi durante i primi due anni di vita. La gravità delle conseguenze varia in base al numero e al tipo di suture coinvolte. Nei casi più gravi, nei quali sono interessate più suture, oltre alla deformità del cranio si sviluppa anche una aumentata pressione al suo interno, nota come ipertensione endocranica. Questa condizione può comportare danni al cervello in evoluzione se non viene correttamente e precocemente diagnosticata e trattata, con impatti potenzialmente devastanti. I danni possono colpire la vista, l’udito e le capacità cognitive. Pertanto, è fondamentale che la patologia venga correttamente diagnosticata ed i bambini possano essere sottoposti al corretto trattamento chirurgico entro i primi anni di vita, quando questo permette di ottenere i migliori risultati sia da un punto di vista funzionale sia estetico.
Tipi Specifici di Craniostenosi e Loro Prevalenza
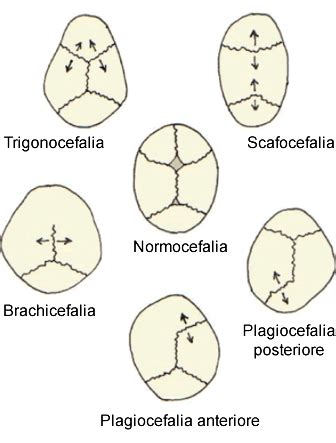
La classificazione delle craniostenosi dipende dalla sutura o dalle suture coinvolte, ognuna delle quali produce una forma cranica caratteristica e ha una propria prevalenza:
- Plagiocefalia Anteriore (Sinostosi Coronarica Unilaterale): Il coinvolgimento di una sola sutura coronarica avviene nella metà dei casi ed è definita plagiocefalia anteriore (sinistra o destra a seconda del lato in cui si è verificata la saldatura). Questa condizione ha una prevalenza di circa 1:10.000 bambini nati e la maggioranza dei casi sono sporadici, con una predominanza per il sesso femminile (rapporto M.F = 1:2).
- Dolicocefalia o Scafocefalia (Sinostosi Sagittale): La prematura fusione delle sutura sagittale viene definita dolicocefalia (testa lunga per il tipico aumento del diametro antero-posteriore) o più comunemente scafocefalia (la conformazione dello scafo della nave). È la più comune craniostenosi con una prevalenza di circa 1:5.000 bambini nati. Gli studi epidemiologici sulle craniostenosi hanno evidenziato una predominanza del sesso maschile per le sinostosi sagittale.
- Trigonocefalia (Sinostosi Metopica): La fusione della sutura metopica avviene nel periodo gestazionale e dà origine alla cosiddetta trigonocefalia. Questa malformazione ha una prevalenza alla nascita di circa 1:15.000 con una predominanza nel sesso maschile (M:F=3:1). Circa il 5-6% dei casi sono famigliari.
- Plagiocefalia Posteriore o Sinostosi Lambdoidea: Questa forma di craniostenosi presenta una prevalenza di 1:15.000 bambini nati con una predominanza nel sesso maschile.
- Sinostosi Complesse o Polisuturali: Queste sono definite come la contemporanea chiusura di più di una sutura e hanno un’incidenza di circa il 5% dei casi non sindromici (cioè non associata a definiti quadri clinici complessi causati da alterazioni genetiche note). Il coinvolgimento di due suture avviene in circa i 2/3 dei casi, e l’interessamento di due o più suture in un terzo dei casi, spesso non interessando suture contigue.
- L’interessamento bilaterale della sutura coronale dà origine ad una conformazione cranica corta, definita brachicefalia con riduzione del diametro anteroposteriore del cranio.
- La chiusura delle due suture lambdoidee comporta una piattezza dell’occipite e viene chiamata pachicefalia.
- La chiusura di più suture, che comporta una crescita “puntuta” del cranio (verso la fontanella anteriore, che chiude per ultima) viene definita oxicefalia.
Eziologia e Fattori di Rischio delle Craniostenosi
La causa delle Craniostenosi è ancora sconosciuta nella maggior parte dei casi. Tuttavia, per alcune forme più gravi, che implicano l’interessamento di più suture, è stata riconosciuta una alterazione genetica ed è nota una trasmissione familiare. In particolare, i geni dei recettori dei fattori di crescita dei fibroblasti (FGFRs) e il gene TWIST sono responsabili delle forme comuni di craniostenosi. I recettori dei fattori di crescita dei fibroblasti (FGFRs) sono costituiti da 4 diverse proteine, codificate da 4 geni diversi. Per quanto riguarda le forme che interessano una sola sutura molta ricerca rimane ancora da fare, sebbene non siano state ancora identificate le alterazioni genetiche è probabile che ve ne siano, in quanto è osservazione frequente una ricomparsa della malformazione nei figli dei soggetti trattatati.
Le craniostenosi possono essere classificate in base alla loro eziologia come primarie, cioè non dovute ad altro disordine, oppure secondarie. Le forme secondarie sono dovute a patologie sottostanti quali metaboliche (come ipertiroidismo, rachitismo, mucopolisaccaridosi), ematologiche (come talassemia, anemia falciforme), malformazioni (incluse oloprosencefalia, microcefalia, encefalocele), teratogeni (come idantoina, retinoidi) o cause iatrogene (come la sindrome da iperdrenaggio in caso di derivazione liquorale).
Spesso le craniostenosi si presentano come anomalie isolate, ma possono anche associarsi ad altre malformazioni o condizioni patologiche all’interno di veri e propri quadri sindromici. Sono note oltre 90 sindromi con craniostenosi associata, di cui circa la metà mostra modalità di trasmissione autosomica dominante o recessiva. In caso di trasmissione autosomica recessiva, due copie del gene sono necessarie per esprimere la malattia, uno ereditato da ciascun genitore, che sono portatori obbligati. Genitori portatori hanno una possibilità su quattro, o 25 per cento, con ogni probabilità la gravidanza, di avere un bambino con craniosinostosi. Maschi e femmine sono ugualmente colpiti. Nella trasmissione autosomica dominante, un gene è necessario per esprimere la malattia e il gene è passato dai genitori ai figli con un rischio 50/50 per ogni gravidanza.
Gli studi epidemiologici sulle craniostenosi hanno evidenziato una predominanza del sesso maschile, in particolare per le sinostosi sagittale e lambdoidea, mentre per le sinostosi coronali si riscontra una predominanza del sesso femminile. Altri fattori di rischio includono la prematurità (età gestazionale < 37 settimane) e il basso peso alla nascita (< 2500 g), che sono riportati quali fattori di rischio. Inoltre, le craniostenosi appaiono associate con l’incremento dell’età paterna (> 40 anni) in particolare per la sinostosi coronale.
Diagnosi e Approccio Terapeutico alla Craniostenosi
La diagnosi delle craniostenosi si basa sull'osservazione clinica e su indagini radiologiche. Nei neonati con questa malattia, il cambiamento nella forma della testa e del viso può essere evidente sin dalla nascita o diventare più marcato con la crescita. Sintomatologicamente, le craniostenosi vanno classificate in compensate e scompensate. Le forme compensate presentano un accrescimento del cervello normale senza apparenti segni di compressione del sistema nervoso centrale. I segni clinici più evidenti in questi casi riguardano principalmente lo scheletro, manifestandosi con deformazione cranica, chiusura precoce della fontanella bregmatica e un ridotto perimetro cranico, oltre ad alterazioni facciali.
Craniosinostosi e il suo trattamento | Ospedale pediatrico di Boston
Le forme scompensate, al contrario, sono palesemente più gravi e determinano una sintomatologia d’ipertensione endocranica, che può instaurarsi in modo subdolo e presentare una sintomatologia invalidante. A causa del rischio di danni neurologici permanenti, la corretta e precoce diagnosi è cruciale.
Il trattamento della malformazione è chirurgico ed ha tre finalità principali:
- Alleviare l’ipertensione endocranica, prevenendo i danni cognitivi e visivi che questa può comportare.
- Permettere che il cranio accolga e “protegga” la crescita del cervello, coprendolo con una volta ossea.
- Cercare di migliorare l’aspetto di questi bambini da un punto di vista estetico, il che è importante per l'integrazione sociale e la qualità della vita.
Deformazioni Craniche Posizionali: Cause e Gestione Iniziale
Le deformazioni craniche di origine posizionale, pur essendo diverse dalle craniostenosi, sono ugualmente importanti da riconoscere e gestire. Esse derivano dalla concomitanza di molteplici cause, che ne possono determinare l’insorgenza o aggravarne la severità.
Le principali cause includono:
- Forze compressive intrauterine: Queste rappresentano la causa più frequente delle deformità craniche posizionali. All’interno dell’utero materno il feto può subire limitazioni negli spostamenti, e trovarsi costretto nella medesima posizione per un periodo di tempo prolungato. La posizione del capo e del collo, a lungo mantenuta, associate alla malleabilità delle ossa, tendono a determinare una deformazione cranica ed uno squilibrio tensivo dei muscoli del collo, in particolare dello sternocleidomastoideo.
- Forze extrauterine compressive: Alcune condizioni anatomiche e/o funzionali osservabili nella madre possono favorire lo sviluppo di deformazioni craniche posizionali, esercitando pressioni esterne sul cranio fetale o neonatale.
- Torcicollo muscolare miogeno: Questa condizione è causata dall’eccessiva ed asimmetrica tensione del muscolo sternocleidomastoideo. Quando uno di questi due muscoli, situati lungo la parte laterale del collo, è teso ed accorciato, la testa del bimbo si mostra piegata in avanti ed inclinata verso il lato interessato dalla tensione, mentre il viso sarà rivolto verso la spalla opposta. A causa di questa squilibrata tensione muscolare, in posizione di riposo la testa tenderà sempre nella stessa posizione, creando una plagiocefalia posizionale, con aree di appiattimento nella regione parieto-occipitale dal lato verso cui la testa è prevalentemente rivolta e, di conseguenza, rigonfiamenti in altre parti del cranio.
Alcune condizioni fisio-patologiche del neonato, come la prematurità, o francamente patologiche, come la sofferenza perinatale, richiedono terapie lunghe ed intensive, che possono determinare una postura obbligata del capo o limitarne in maniera importante la motilità, contribuendo all'insorgenza o al peggioramento delle deformità posizionali.
Le anomalie morfologiche del cranio determinate esclusivamente da eventi meccanici relativi al periodo espulsivo tendono a regredire spontaneamente nel giro di alcune settimane ed usualmente richiedono soltanto un programma di terapia fisica. Tuttavia, l’asimmetria del capo è spesso di modesta entità alla nascita, può sfuggire alla considerazione del pediatra e passare inosservata ai genitori. Il neonato con deformazione cranica posizionale mostra spesso di preferire il decubito supino dal lato appiattito dalla costrizione uterina. Se il piccolo non viene precocemente condizionato a privilegiare decubiti alternativi, l’asimmetria cranica manifesterà un’ineluttabile progressione peggiorativa. L'intervento precoce, spesso con semplici modifiche posturali e fisioterapia, è fondamentale per prevenire l'aggravamento e favorire la risoluzione.
La Natimortalità: Definizione, Incidenza e Cause
La natimortalità è un evento tragico che comporta la morte del feto prima o durante il parto. Negli Stati Uniti, la natimortalità è definita come la morte fetale a ≥ 20 settimane di gestazione. L'Organizzazione Mondiale della Sanità, invece, definisce la natimortalità come morte fetale dopo 28 settimane. A livello globale, ci sono quasi 2 milioni di natimortalità in tutto il mondo ogni anno (1). Una pregressa morte fetale aumenta il rischio di natimortalità nelle gravidanze successive, sottolineando l'importanza di un'attenta anamnesi e gestione in caso di eventi pregressi.
L'eziologia della natimortalità è complessa e multifattoriale. La morte fetale durante una gravidanza avanzata può avere cause materne, placentari, fetali oppure genetiche. Spesso, nonostante indagini approfondite, non è possibile determinarne la causa specifica. Tuttavia, l'identificazione di fattori di rischio e cause potenziali è cruciale per la prevenzione e per il supporto alle famiglie. Tra le complicanze che possono verificarsi, se un feto muore in tarda gravidanza o vicino al termine ma rimane nella cavità uterina per settimane, si può verificare coagulopatia da consumo o anche coagulazione intravascolare disseminata nella madre, una condizione grave che richiede gestione medica immediata.
Diagnosi e Gestione della Natimortalità
La diagnosi di natimortalità è clinica, basandosi sull'assenza di attività cardiaca fetale. Una volta accertata, il passo successivo è determinare la causa, il che richiede una serie di test e valutazioni. I test per identificare la causa della natimortalità possono comprendere i seguenti:
- Esame generale del feto nato morto: Include l'aspetto fisico, il peso, la lunghezza e la circonferenza cranica (1).
- Autopsia fetale, cariotipo e microarray: Queste analisi genetiche e patologiche sono fondamentali per identificare anomalie cromosomiche o strutturali che potrebbero aver causato la morte fetale.
- Esame della placenta: La placenta è un organo vitale e le sue anomalie possono essere una causa significativa di natimortalità.
- Test materni:
- Emocromo con formula materno (per evidenziare l'anemia o la leucocitosi).
- Test di Kleihauer-Betke (per rilevare emorragia feto-materna).
- Screening diretto per disturbi trombotici acquisiti, inclusi test per anticorpi antifosfolipidi (lupus anticoagulante, anticardiolipina [IgG e IgM], anti-beta2 glicoproteina I [IgG e IgM]).
- Ormone stimolante la tiroide, e se anomalo, T4 libera (tiroxina) per valutare la funzione tiroidea materna.
- Test del diabete (HbA1C) per valutare il controllo glicemico materno.
- TORCH test (toxoplasmosi [con IgG e IgM], altri agenti patogeni [p. es., parvovirus B19 umano, virus varicella-zoster], rosolia, cytomegalovirus, herpes simplex) per infezioni.
- Test sierologici reaginici (Rapid Plasma Reagin [RPR]) per la sifilide.
- Test tossicologici per identificare l'esposizione a sostanze nocive.
L'esame per la trombofilia ereditaria è controverso e non è raccomandato di routine. L’associazione tra natimortalità e trombofilia ereditaria non è chiara ma non sembra essere forte, fatta eccezione per la possibile mutazione del fattore V di Leiden. I test (p. es., per il fattore V di Leiden) possono essere considerati quando vengono rilevate gravi anomalie nella placenta, si verifica una restrizione della crescita intrauterina o se la donna ha un’anamnesi personale o familiare di malattie tromboemboliche (1).
Riferimento generale: 1. American College of Obstetricians and Gynecologists, Society for Maternal-Fetal Medicine: Management of stillbirth. Obstetric Care Consensus No. 10, 2020. Confermato nel 2021.
La gestione della natimortalità implica lo svuotamento uterino se necessario. Lo svuotamento uterino si può verificare spontaneamente. In caso contrario, lo svuotamento deve essere fatto utilizzando farmaci (p. es., ossitocina) o una procedura chirurgica (p. es., la dilatazione e lo svuotamento [D & E], preceduti da dilatatori osmotici pre abortivi per preparare la cervice, con o senza misoprostolo), a seconda dell’età gestazionale. Dopo l’espulsione dei prodotti del concepimento può essere necessario un curettage per rimuovere ogni frammento placentare ritenuto. I frammenti hanno un numero maggiore di probabilità di rimanere quando la natimortalità avviene in un periodo molto iniziale della gravidanza. Se si sviluppa una coagulazione intravascolare disseminata, la coagulopatia deve essere gestita prontamente e in modo aggressivo, sostituendo il sangue o i prodotti del sangue, secondo necessità.
Craniosinostosi e il suo trattamento | Ospedale pediatrico di Boston
Il trattamento nel post partum è simile a quello per il parto di un nato vivo, ma con un'enfasi particolare sul supporto emotivo. I genitori generalmente si sentono estremamente afflitti e richiedono un supporto emozionale e talvolta richiedono una consulenza psicologica formale. Con le pazienti devono essere discussi i rischi, che sono relativi alle cause presunte, per le future gravidanze.
L'Intersezione tra Malformazioni Craniche e Natimortalità
L'interconnessione tra malformazioni craniche e natimortalità è un aspetto critico. Sebbene la maggior parte delle malformazioni craniche posizionali non sia direttamente correlata alla natimortalità, le forme più gravi di craniostenosi e altre malformazioni cerebrali o craniche complesse possono avere un impatto significativo sulla sopravvivenza fetale. Le malformazioni (come oloprosencefalia, microcefalia, encefalocele) sono state identificate come cause di craniostenosi secondaria, indicando una correlazione tra difetti strutturali cerebrali e cranici e la chiusura anomala delle suture.
In particolare, alcune malformazioni cerebrali e craniche sono per loro natura incompatibili con la vita. L'anencefalia, ad esempio, è una condizione fatale a causa delle gravi malformazioni cerebrali che comporta. Questo difetto è causato dalla mancata chiusura del tubo neurale anteriore che deve avvenire entro la 4a settimana di gestazione per poi formare la colonna vertebrale e il cervello. La chiusura del tubo neurale anteriore, definita “chiusura rostrale”, si verifica normalmente all’inizio dello sviluppo cerebrale; eventuali anomalie hanno perciò sempre gravi conseguenze, come l’anencefalia. In questi casi, la diagnosi prenatale di una malformazione così grave può portare alla natimortalità, sia come esito naturale della condizione, sia in relazione a scelte mediche e familiari. La natimortalità, per definizione, comporta la morte del feto, e le cause fetali o genetiche, come malformazioni complesse e incompatibili con la vita, ne costituiscono una componente significativa. La presenza di tali malformazioni può essere un fattore diretto o indiretto che contribuisce alla morte intrauterina del feto, rientrando nell'ampia categoria delle cause fetali della natimortalità.
Malformazioni Congenite: Una Prospettiva più Ampia di Cause, Tipi e Prevenzione
Le malformazioni congenite, o anomalie congenite, sono difetti strutturali o funzionali che si sviluppano durante la vita intrauterina e possono essere rilevati prima della nascita, alla nascita o in un momento successivo. Le malformazioni congenite hanno un’incidenza del 2-3% nella popolazione generale e nell’80-90% dei casi si verificano in coppie che non presentavano un rischio specifico. Le anomalie strutturali, ovvero le anomalie nello sviluppo delle parti del corpo, possono variare enormemente in tipologia e gravità.
Le cause scatenanti sono varie e spesso complesse:
- Cause genetiche: Molte malformazioni sono ereditate o derivano da nuove mutazioni genetiche o cromosomiche. La fenilchetonuria (PKU), ad esempio, è una malattia ereditaria che porta ad un accumulo di fenilalanina nel corpo.
- Cause ambientali: Esposizione a teratogeni (sostanze chimiche, farmaci, infezioni) durante la gravidanza.
- Fattori multifattoriali: La maggior parte dei difetti alla nascita sono multifattoriali, ovvero causati da una combinazione di cause genetiche e ambientali.
Tra le malformazioni congenite più diffuse, oltre a quelle craniche già menzionate, si annoverano:
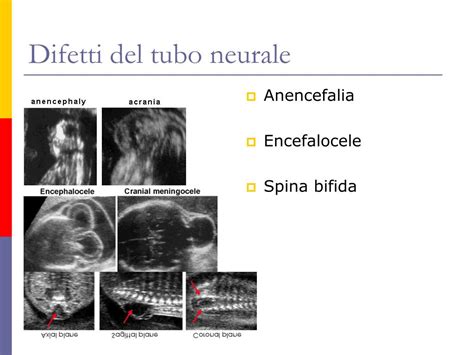
- Difetti del tubo neurale: Questi sono un tipo di difetto alla nascita che colpisce il cervello o il midollo spinale. "Difetti del tubo neurale" è un termine generico che comprende diverse malformazioni, due delle più comuni sono la spina bifida e l’anencefalia. Sebbene non vi sia una cura per i difetti del tubo neurale, le donne incinta dovrebbero assumere acido folico non appena ottenuta la diagnosi di gravidanza per diminuire la probabilità che il loro bambino sviluppi un difetto del tubo neurale. La mancata assunzione della giusta quantità di acido folico può essere un fattore di rischio di disturbo genetico. La spina bifida è solitamente rilevabile mediante ecografia ed altri test prenatali.
- Difetti cardiaci congeniti (CHD): Costituiscono le malformazioni alla nascita frequenti. Questi difetti influenzano il flusso di sangue nel cuore e, di conseguenza, in tutto il corpo. I CHD variano in gravità e possono causare molti problemi per tutta la vita. Per correggere un difetto cardiaco congenito è solitamente necessaria la chirurgia.
- Palatoschisi o labbro leporino: Un bambino nato con palatoschisi o labbro leporino richiederà di solito un intervento chirurgico per riparare la condizione. Queste condizioni indicate fanno parte dei difetti alla nascita che possono derivare da insufficiente acido folico o altri fattori.
- Gastroschisi e Onfalocele: La gastroschisi è un difetto congenito consistente nella presenza di un buco nell’addome che permette agli organi di sporgere dal corpo. L’onfalocele viene spesso rilevato durante il secondo e il terzo trimestre di gestazione attraverso l’ecografia.
- Ipospadia: Questa condizione colpisce i neonati maschi ed è un difetto in cui l’uretra non esce dal pene nella giusta posizione.
- Malformazioni degli arti: Se tuo figlio è nato con una malformazione degli arti, è consigliabile rivolgersi a un legale con esperienza in malformazioni congenite.
- Microcefalia: Si tratta di una condizione di insufficiente crescita della testa del feto che può essere diagnosticata durante la gravidanza. Questa condizione determina il sottosviluppo del cranio e del cervello del feto e di solito è fatale.
- Anoftalmia e Microftalmia: Sono caratterizzate, rispettivamente, dall’assenza di un occhio e dalla presenza di un occhio più piccolo.
- Encefalocele: È un difetto alla nascita caratterizzato dalla sporgenza di cervello e cranio. Può portare a disabilità a lungo termine. Può essere diagnosticato mediante ecografia durante la gravidanza: si evidenzia la presenza di un sacco cistico, attaccato al cranio.
Le infezioni in gravidanza rappresentano una causa biologica rilevante nell’eziologia di alcune malformazioni fetali. Il danno embrionale non si manifesta nella totalità dei casi e la gravità è strettamente correlata al periodo di esposizione. Generalmente tanto più precoce è l’infezione, tanto maggiori sono le probabilità di causare un’embriopatia e tanto maggiori sono in termini di gravità gli effetti fetali. La consulenza teratologica per esposizione a farmaci in gravidanza mira a valutare i possibili rischi fetali in relazione al tipo, alla durata e all’epoca di esposizione.
Diagnosi Prenatale e Prevenzione delle Malformazioni Congenite
La diagnosi precoce è fondamentale per la gestione delle malformazioni congenite. In determinati casi le malformazioni genetiche/congenite del feto sono sospettabili e diagnosticabili in gravidanza attraverso vari metodi di screening e diagnostica prenatale.
- Screening del primo trimestre: Ad esempio, la misurazione della translucenza nucale, eseguibile tra la 11a e la 13a settimana gestazionale, permette, in combinazione con il dosaggio ematico di alcuni marcatori biochimici, di valutare il rischio di alcune cromosomopatie (es. Sindrome di Down). La fisiopatologia dell’aumento della translucenza nucale non è ancora chiara, tuttavia una compromissione dell’attività cardiaca sembra essere un meccanismo importante. Altre cause possibili sono la congestione venosa a livello della testa e del collo, l’alterata composizione della matrice extracellulare, un anomalo o mancato sviluppo del sistema linfatico, il malfunzionamento del drenaggio linfatico, anemia o ipoproteinemia fetale ed infezioni congenite.
- Diagnosi prenatale non invasiva (NIPT): Eseguita su sangue materno analizza il rischio delle trisomie fetali più comuni e aneuploidie X, Y in gravidanze dalla decima settimana in poi. L’esame ha dimostrato un’attendibilità superiore al 99% nel determinare il rischio di trisomia 21 (Sindrome di Down) e rispettivamente del 98% e 80% per le trisomie 18 e 13 (Sindrome di Edwards e Sindrome di Patau), con un tasso di falsi positivi inferiore allo 0.1% in tutti e tre i casi.
- Ecografia morfologica: Nel secondo trimestre di gestazione la valutazione ecografica permette l’identificazione di molte malformazioni congenite attraverso lo studio accurato dell’anatomia fetale. Tra le malformazioni congenite più frequenti diagnosticabili in epoca prenatale vi sono le cardiopatie congenite, le malformazioni cerebrali, le malformazioni intestinali e renali. Anche l'indagine ecografica può identificare anomalie nello sviluppo del cranio del bambino. La mancata esecuzione di test diagnostici adeguati o l’errore nella interpretazione delle immagini dell’esame sono ipotesi di colpa medica.
Non essendo ancora noto con certezza ciò che causa molte malformazioni, non esistono a oggi sistemi sicuri di prevenzione oltre a specifiche raccomandazioni. Cosa si può fare, quindi, per prevenire le malformazioni congenite? Beh, innanzitutto condurre una vita sana, con particolare attenzione se si programma una gravidanza. Lo strumento preventivo più efficace a oggi è l’assunzione di integratori vitaminici prima e durante le prime fasi della gravidanza per fornire un supplemento di acido folico alle donne incinte o che desiderano una gravidanza. Le madri devono essere consapevoli della necessità di assumere acido folico e dei farmaci che in gravidanza vanno evitati. Altre misure preventive includono il controllo dello stato vaccinale, in particolare contro la rosolia, una malattia che può provocare un aborto spontaneo se contratta durante la gravidanza.
In alcuni casi, alla madre potrebbero essere stati prescritti durante la gravidanza determinati farmaci dei cui rischi potrebbe non essere stata pienamente informata. Diversi farmaci sono stati collegati all’insorgenza di condizioni cardiache nei neonati. L’assunzione di questi farmaci durante la gravidanza è un fattore di rischio di malattie cardiache. Se la condizione viene scoperta durante la gravidanza, è spesso troppo tardi per effettuare un trattamento.
Aspetti Legali e il Diritto al Risarcimento
Tutti facciamo affidamento sul fatto che i professionisti medici trattino adeguatamente i nostri problemi di salute e rispondano in modo adeguato ad emergenze mediche. Quando si parla di errori medici, spesso le persone si riferiscono agli errori chirurgici. Anche se è vero che la maggior parte delle negligenze mediche rientrano nella categoria chirurgica, gli errori medici possono verificarsi in molti modi diversi. Una serie di errori, ad esempio, si verificano prima della nascita e durante la gravidanza.
Venire a conoscenza del fatto che il proprio figlio sia affetto da una malformazione congenita / genetica è un evento straziante e stravolgente anche per la famiglia più risoluta. Oltre al costo emotivo, il trattamento della maggior parte delle malformazioni congenite / genetiche comporta trattamenti a vita e, a volte, molti interventi chirurgici. Le spese per l’assistenza, inoltre, possono durare tutta la vita del bambino e raggiungere importi molto elevati. Chi dovrebbe sostenere questi costi? La risposta è ovvia. Se si può stabilire che un medico o un ospedale sia in qualche modo legalmente responsabile, questo peso finanziario deve essere tolto alla famiglia del bambino ed attribuito al medico e/o all’ospedale.
La mancata informazione alla gestante circa la possibilità di diagnosi prenatali o sui rischi di determinate esposizioni determina una violazione del diritto all’interruzione volontaria della gravidanza, qualora tale opzione fosse stata considerata. La mancata esecuzione di test diagnostici adeguati o l’errore nella interpretazione delle immagini dell’esame, come nell'ecografia per l'anencefalia o la spina bifida, sono ipotesi di colpa medica. Analogamente, la mancata prescrizione del giusto quantitativo di acido folico può determinare il diritto al risarcimento del danno, data la sua importanza cruciale nella prevenzione di difetti del tubo neurale. Se si sospetta che un farmaco sia responsabile delle condizioni cardiache del bambino, è consigliabile contattare un avvocato con esperienza nel settore per conoscere le proprie opzioni legali.
Una volta che è stata fatta una diagnosi medica, è consigliabile contattare immediatamente un legale esperto, poiché un’adeguata indagine da parte dell’avvocato necessiterà di un po’ di tempo per essere effettuata correttamente. A causa della grande quantità di conoscenze tecniche richieste nella trattazione di casi di malformazione genetica / congenita, rivolgersi ad uno staff di professionisti con esperienza nel settore è fondamentale. I professionisti dello studio legale Stefano Gallo, come riportato, potranno analizzare attentamente il tuo caso gratuitamente. Gli avvocati di Dannidaparto.legal hanno un’esperienza pluriennale in casi di danni occorsi al neonato e alla madre a causa errori medici. Il Dr. Sebastiano Bianca, medico, specialista in Genetica Medica e Dottore di Ricerca in Malattie Genetiche dell’Età Evolutiva, cofondatore di BGenetica e responsabile dei percorsi dedicati alla genetica riproduttiva e alla neurogenetica, offre un'importante competenza in questo ambito specialistico.