Le malformazioni polmonari congenite rappresentano anomalie significative dello sviluppo dei polmoni che si manifestano quando il bambino sta crescendo nell’utero materno. Talvolta, durante questo delicato processo di formazione, alcuni meccanismi si alterano o un "ingranaggio" non funziona correttamente, impedendo ai polmoni di svilupparsi in modo adeguato. Questi organi vitali sono suddivisi in lobi, con tre lobi nel polmone destro e due nel polmone sinistro. Ogni lobo costituisce un'unità anatomica ben definita, dotata di arteria e vena per l’apporto sanguigno e di un bronco da cui riceve l’aria. Le malformazioni polmonari possono essere estese a un solo lobo o a parte di esso, oppure possono interessare più di un lobo, e talvolta essere bilaterali.
La diagnosi di queste anomalie avviene generalmente in epoca prenatale, tra la diciottesima e la ventesima settimana di gestazione. Fortunatamente, nella maggior parte dei casi, queste malformazioni non compromettono il normale sviluppo del feto e non determinano problemi respiratori alla nascita. Tuttavia, come si vedrà, è fondamentale riconoscerle e gestirle correttamente per evitare lo sviluppo di complicanze, talvolta anche gravi, quali infezioni respiratorie, il collasso del polmone o un'evoluzione maligna. L'obiettivo principale, se trattate correttamente, è che non abbiano conseguenze significative sulla qualità di vita dei bambini affetti.
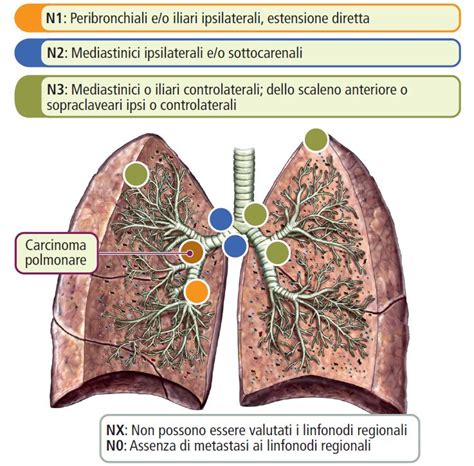
Classificazione delle Malformazioni Polmonari Congenite
Esistono diverse tipologie di malformazioni polmonari congenite, ognuna con caratteristiche specifiche. Le più comuni includono la Malformazione Adenomatoide Cistica Congenita del Polmone (MACCP o CPAM), il sequestro polmonare, l'enfisema lobare congenito e la cisti broncogena.
Malformazione Adenomatoide Cistica Congenita del Polmone (MACCP/CPAM)
La Malformazione Adenomatoide Cistica Congenita del Polmone (MACCP), spesso abbreviata come CPAM (Congenital Pulmonary Airway Malformation) o CCAM (Congenital Cystic Adenomatoid Malformation), è una malformazione congenita benigna del polmone. È caratterizzata da uno sviluppo anomalo dei bronchioli terminali, i quali proliferano in maniera anomala all'inizio dello sviluppo embrionale, generando cisti intraparenchimali di varie dimensioni. Questo processo porta alla formazione di una massa cistica o solida di tessuto polmonare, con la soppressione dello sviluppo degli alveoli.
La malformazione è rara, con una frequenza riportata che va da 1:15.000 a 1:35.000 nati vivi, e si ritiene che si origini verso la quinta settimana di gravidanza. Ad oggi, i fattori responsabili della sua formazione non sono ben noti, e le cause della malattia adenomatoide cistica congenita del polmone sono sconosciute. Si presenta come una massa occupante spazio, di solito unilaterale in più del 95% dei casi, e solitamente coinvolge un lobo o un segmento del polmone. Nella sua evoluzione, può determinare dislocazione del mediastino e del cuore. L'ecostruttura distingue due forme: una forma multicistica caratterizzata dalla presenza di multiple cisti di varia grandezza (tipo I e II) e una forma microcistica (tipo III) corrispondente alla variante solida.
La classificazione di Stocker distingue tre principali tipi di MACCP:
- Tipo I: Ha una buona prognosi ed è presente in circa il 50% dei casi. Le cisti sembrano comunicare con il normale parenchima polmonare e possono essere circondate da cisti più piccole. Questo tipo è caratterizzato da cisti di dimensioni maggiori, spesso singole o multiple, di diametro superiore a 2 cm.
- Tipo II: Si verifica in circa il 40% dei casi ed è costituito da numerose piccole cisti (minori di 2,0 cm) mescolate con aree di aumentata ecogenicità all'ecografia. La prognosi dipende dalla gravità della lesione e dalla sua associazione con altre malformazioni fetali. Esiste un'associazione tra anomalie congenite e CCAM di tipo II, le principali delle quali sono genitourinarie, come la disgenesia o l'agenesia renale, e cardiache, come la tetralogia di Fallot e il tronco arterioso; altri includono l'ernia diaframmatica e le anomalie muscoloscheletriche.
- Tipo III: È una massa solida che contiene cisti molto piccole (minori di 5,0 mm), caratterizzata da aree di aumentata ecogenicità all'ecografia. Questo tipo è associato a una prognosi più riservata, in quanto può determinare dislocazione del mediastino e alterazione della normale ecostruttura dei campi polmonari, con il potenziale di peggiorare la prognosi determinando l’insorgenza di idrope fetale.
Uno studio importante, considerato una delle più grandi coorti di casi (Chen Y et al., 2021), conclude che le CCAM microcistiche e macrocistiche presentano entrambe decorsi e prognosi perinatali simili.
Sequestro Polmonare
Il sequestro polmonare è un'altra forma di malformazione polmonare congenita. Si tratta di una massa di tessuto polmonare non funzionante che non comunica con l’albero tracheobronchiale e che riceve la vascolarizzazione da un vaso anomalo, generalmente proveniente dall’aorta toracica o addominale. In altre parole, una porzione del polmone non riceve l’aria e non è quindi utile alla respirazione. Questa porzione di tessuto polmonare non funzionante è irrorata da un vaso arterioso anomalo proveniente dall’aorta. È una lesione monolaterale, localizzata nella maggior parte dei casi al lobo inferiore del polmone sinistro.
Il sequestro può essere:
- Intralobare: localizzato all’interno di un lobo polmonare (75% dei casi).
- Extralobare: localizzato all’esterno (25% dei casi). Questa forma è più rara ed è quella classicamente diagnosticata in epoca prenatale; nel 90% dei casi è sopra-diaframmatica, mentre nel 10% è sotto-diaframmatica. Si presenta come una lesione iperecogena, localizzata più frequentemente al lobo inferiore del polmone sinistro, più raramente in sede sottodiaframmatica.
Per il sequestro polmonare, deve essere valutata l'eventuale presenza di idrotorace oppure di idrope.
Enfisema Lobare Congenito
L'enfisema lobare congenito si manifesta come l'iperespansione di un lobo polmonare anatomicamente normale. Questa condizione, pur coinvolgendo un lobo senza alterazioni strutturali intrinseche evidenti a livello microscopico come nelle CCAM, può comunque causare compressione delle strutture adiacenti e problemi respiratori.
Cisti Broncogena
Una cisti broncogena è una cisti dotata di una parete simile a quella bronchiale, che può svilupparsi in qualsiasi parte del torace, inclusi i polmoni, il mediastino o anche al di sotto del diaframma. Queste cisti possono rimanere asintomatiche per lungo tempo o causare sintomi per compressione o infezione.
Diagnosi Prenatale e Monitoraggio della Gravidanza
La prima difficoltà nella gestione delle malformazioni polmonari congenite consiste nell'effettuare una corretta diagnosi, poiché spesso sono asintomatiche sia in epoca fetale che neonatale. Il bambino può apparire quindi perfettamente sano durante la gravidanza e subito dopo il parto. Attualmente, la diagnosi mediante ecografia prenatale è sempre più frequente ed è possibile dal secondo trimestre di gestazione, solitamente nel corso dell'ecografia di screening del secondo trimestre, la cosiddetta ecografia "morfologica", tra la diciottesima e la ventesima settimana di gravidanza.
L'ecografia del secondo trimestre può dimostrare la presenza di alcune cisti nel polmone o la presenza di una zona iperecogena (più chiara) al suo interno. Non è indicata l’esecuzione del cariotipo e dell’ecocardiografia fetale, in assenza di altre anomalie all’ecografia. Tuttavia, tramite l’ecografia, non è possibile determinare con sicurezza di quale tipo di malformazione si tratti.
In caso di sospetto ecografico prenatale di MACCP o sequestro polmonare, presso centri specializzati, viene solitamente consigliato di eseguire una Risonanza Magnetica (RM) fetale. Questo esame radiologico non è dannoso né per la mamma né per il feto, in quanto non espone a radiazioni ionizzanti. La risonanza magnetica non è nota avere effetti dannosi sul feto umano per esposizione in modo sporadico e per alcune decine di minuti. La RM permette una migliore definizione della malformazione rispetto all’ecografia, anche se non consente comunque una diagnosi definitiva. L'ecografia prenatale non permette di definire con certezza né l’evoluzione fetale, né la prognosi post-natale dei singoli casi.
Malformazioni fetali - Casi clinici
Durante la gravidanza, il compito dell'équipe medica, a seguito della diagnosi, è quello di fornire ai genitori tutte le informazioni necessarie per prendere una decisione consapevole e definire insieme un percorso di cura condiviso, accompagnandoli nelle varie fasi: dalla prenatale a quella postnatale. Consigliamo innanzi tutto di assumere informazioni sulla malattia solo da centri esperti e di eccellenza.
La malformazione adenomatoide cistica del polmone (CAM) ha una storia naturale variabile che può evolvere in idrope o regredire. Non sono disponibili criteri certi per determinare quali lesioni crescerebbero e svilupperebbero idrope rispetto a quelle la cui crescita si stabilizzerebbe o regredirebbe. Le cisti possono crescere progressivamente fino alle 26 settimane. Un'involuzione spontanea è possibile nel 20% circa dei casi e la si può dimostrare con l'ecografia. La completa e reale scomparsa deve essere comunque documentata con una tomografia computerizzata del torace a partire dal terzo mese di vita dopo la nascita. L'iperecogenicità del polmone (o le cisti) si riduce durante la gravidanza fino a scomparire in alcuni casi.
Per calcolare il rischio di sviluppare l’idrope fetale, è stato proposto l’utilizzo del rapporto volumetrico CAM Volume Ratio (CVR), cioè il rapporto tra il volume della MACP e la circonferenza cranica (Crombleholme TM et al., 2002). Il volume della MACP viene calcolato moltiplicando i tre diametri della massa x 0,52. I feti con CVR inferiore o uguale a 1,6 sono considerati a basso rischio di sviluppo di idrope, e quelli con CVR maggiore di 1,6 sono considerati a rischio aumentato per lo sviluppo dell'idrope. La CVR è un utile indicatore ecografico dei feti a rischio di idrope che richiedono un'attenta osservazione ecografica e un possibile intervento fetale (Tran Hong et al., 2008). In caso di CPAM microcistiche e/o un CVR >1,6, è stato valutato l'effetto dell'uso prenatale di un singolo ciclo di steroidi. È stata osservata una riduzione della lesione nel 61% dei casi e l'idrope fetale si è risolta nel 78% (Curran PF et al., 2010). I dati disponibili in letteratura dimostrano che il suo utilizzo ha contribuito in modo significativo al miglioramento della prognosi fetale e perinatale, ma nonostante i risultati positivi della terapia con corticosteroidi nel trattamento dei feti affetti da CPAM ad alto rischio, è ancora necessario effettuare ulteriori studi sull’argomento (Macedo I., et al. 2022).
La gravidanza dovrebbe inizialmente essere gestita presso un centro di III livello con ecografie seriali. La valutazione ecografica deve essere eseguita settimanalmente per valutare il volume della CCAM, e la CVR può identificare i primi segni di idrope fetale. Se sono presenti cisti dominanti, anche con un CVR <1,6, esiste il rischio di crescita acuta della cisti e sviluppo di idrope fetale (Rolo LC et al., 2022).
Gestione delle Complicanze Fetali e del Parto
Solo una minoranza di casi può presentare sintomi gravi durante la vita fetale. La presenza della malformazione può causare, infatti, l’alterazione della normale disposizione degli organi nel torace e la compressione del cuore e dei vasi sanguigni principali. La conseguenza più grave di ciò è rappresentata da una compromissione della circolazione con conseguente idrope fetale, ossia l’accumulo di liquido nei tessuti fetali (edema del tessuto sottocutaneo, versamento toracico e pericardico, ascite). In caso di insorgenza di idrope fetale, le probabilità di sopravvivenza del feto sono purtroppo basse.
Studi dimostrano che, senza il trattamento dei feti con CPAM ad alto rischio, il rischio di sviluppare idrope, che è un importante predittore di mortalità fetale, aumentava fino al 100% (Crombleholme TM et al., 2002; Leblanc C. et al., 2017). Esiste però, in pochi centri altamente specializzati come l'IRCCS Ca’ Granda - Ospedale Maggiore Policlinico di Milano, la possibilità di eseguire trattamenti fetali che mirano a migliorare le condizioni cliniche del feto aumentandone la probabilità di sopravvivenza. L’obiettivo di questi trattamenti è portare il feto il più possibile vicino al termine della gravidanza e poterlo poi trattare definitivamente dopo la nascita. La possibilità di eseguire questi trattamenti fa sì che l’indicazione all’interruzione terapeutica della gravidanza possa essere riservata solo a casi di particolare compromissione del feto o in presenza di altre gravi patologie o malformazioni associate.
Per le malformazioni isolate e asintomatiche per il feto, non c’è l’indicazione a eseguire ulteriori approfondimenti in utero, e il parto potrà avvenire per via naturale. È però indicato eseguire comunque il parto in un centro di terzo livello. Infatti, anche se nella maggior parte dei casi un feto che si mantiene asintomatico durante la gravidanza non ha problemi durante e subito dopo il parto, è tuttavia possibile che presenti delle difficoltà respiratorie alla nascita. Quindi è opportuno che questi bambini nascano in centri che abbiano sempre a disposizione le competenze e gli strumenti adeguati alla gestione di eventuali urgenze respiratorie neonatali. È bene che il bambino nasca a termine di gravidanza, per garantire il completo sviluppo polmonare, e da parto spontaneo, se possibile.
Il distress respiratorio può essere causato da una malattia adenomatoide cistica congenita del polmone di dimensioni importanti o può dipendere dalla presenza di un versamento pleurico che impedisce alla parte di polmone sano di espandersi e, quindi, di ventilare. In caso di difficoltà respiratoria, il bambino verrà portato in Neonatologia e riceverà assistenza ventilatoria. Meno del 10% di questi casi richiede un trattamento chirurgico alla nascita, per la presenza immediata di sintomi respiratori, anche modesti.
Diagnosi Postnatale e Pianificazione dell'Intervento Chirurgico
Se, come accade nella maggior parte dei casi, la gravidanza è decorsa regolarmente e il bambino è asintomatico alla nascita, nei primi giorni di vita viene sottoposto ad una Risonanza Magnetica (RM) del torace in sonno spontaneo. La RM, come già detto, non espone a radiazioni ionizzanti e non è quindi dannosa. Inoltre, viene eseguita durante il sonno del neonato, senza necessità di somministrare farmaci per sedarlo, né mezzo di contrasto. Fino a pochi anni fa si era soliti eseguire una Tomografia Computerizzata (TC) alla nascita per la conferma diagnostica e una seconda TC pre-operatoria per definire nel dettaglio l’anatomia della malformazione. Fortunatamente oggi, grazie alla RM in sonno spontaneo, è possibile limitare l’utilizzo della TC a un solo esame in prossimità dell’intervento chirurgico.
Nel 2/3 dei casi il bambino nasce e, come ci si attendeva, non ha alcun sintomo respiratorio. Viene dunque eseguita una semplice radiografia. Qualora questa non consenta di visualizzare alcuna lesione polmonare (evenienza frequente), consigliamo di portare il bambino tranquillamente a casa. Effettuerà un primo controllo presso l'ambulatorio di Chirurgia Neonatale a circa 1 settimana di vita e successivi regolari controlli del pediatra curante. Alcuni bambini non manifestano sintomi fino ai 4-6 mesi. Nel caso di bambino asintomatico, anche quando la malformazione non era più visibile nell’ultimo periodo della gravidanza, verrà programmata una TAC del torace con mezzo di contrasto ai 3 mesi di vita.
Secondo l'esperienza del centro di riferimento, una diagnosi precoce e sufficientemente attendibile è molto utile, poiché permette di stabilire un adeguato percorso assistenziale nei mesi che precedono l’intervento chirurgico. In particolare, consente di prevedere il potenziale rischio di infezioni respiratorie cui il bambino può incorrere e di stabilire, quindi, l'indicazione ad eseguire una profilassi antibiotica quotidiana che prevenga tale complicanza. La malformazione va asportata anche nelle forme che non presentano sintomi. Infatti, il tessuto polmonare non funzionante può causare accumulo di secrezioni e quindi l’insorgere di infezioni. Oltre alle complicanze neoplastiche, queste malformazioni possono predisporre allo sviluppo di infezioni respiratorie con conseguenze anche gravi.
Se tutto procede come previsto, l’intervento viene programmato entro il 5°-6° mese di vita. Si aspetta qualche mese per permettere al bambino di crescere e raggiungere un peso adeguato, ma si cerca comunque di operarlo precocemente, sia per evitare eventuali infezioni respiratorie, sia affinché la parte di polmone che resta dopo l’intervento abbia più tempo per crescere e sostituire la funzione della porzione asportata. Poco prima dell’intervento, viene eseguita una TC del torace. Questo esame radiologico, seppure esponga a radiazioni ionizzanti e richieda che il bambino venga sedato, rappresenta ancora un passaggio fondamentale per una precisa definizione della malformazione, delle vie aeree e dei vasi sanguigni. È quindi necessaria per una corretta preparazione all’intervento chirurgico, agendo come ausilio ai chirurghi.
L'Intervento Chirurgico: Tecniche e Procedure
L’operazione ha l’obiettivo di asportare il tessuto polmonare sede della malattia, risparmiando il tessuto sano. Una volta asportato il tessuto polmonare anomalo, il problema del bambino è risolto.A seconda dell’estensione e della disposizione della malformazione si può decidere di asportare un lobo polmonare (lobectomia), parte di esso (segmentectomia) o rimuovere la malformazione seguendone il profilo (enucleazione/resezione atipica). Nel caso di un sequestro polmonare extralobare, il tessuto polmonare non è interessato e perciò si rimuove solo la malformazione (sequestrectomia).
La lobectomia è l’intervento eseguito più frequentemente nei centri di riferimento. Questo tipo di intervento permette di rispettare l’anatomia del polmone e di non correre il rischio di lasciare in sede porzioni di polmone malato. Anche se potrebbe sembrare un intervento più aggressivo, non è gravato da un peggior risultato in termini di funzionalità respiratoria. Il lobo o i lobi polmonari residui, infatti, sono in grado di espandersi e sostituire completamente il lobo asportato. La segmentectomia o la resezione atipica, invece, sono riservate ai casi in cui la malformazione sia diffusa a più lobi polmonari o ad entrambi i polmoni.
La tecnica toracoscopica rappresenta un importante passo avanti nell’approccio terapeutico ai piccoli pazienti con patologie congenite, riducendo significativamente l’impatto fisico degli interventi di chirurgia maggiore. Dal 2012, presso l'U.O.C. di Chirurgia Pediatrica dell’IRCCS Ca’ Granda - Ospedale Maggiore Policlinico di Milano e della S.C., è nato un rapporto di stretta collaborazione con il Prof. S. Rothenberg, Direttore della Chirurgia Pediatrica del Rocky Mountain Hospital for Children di Denver (USA), universalmente riconosciuto a livello internazionale per la sua attività in ambito chirurgico mini-invasivo, soprattutto toracoscopico. La tecnica toracoscopica richiede strumentari specifici, un team dedicato e la corretta preparazione ed esperienza, che nel centro italiano è stata raggiunta anche grazie alla collaborazione e agli insegnamenti del Prof. S. Rothenberg. In questi anni, tutti gli interventi per malformazione polmonare congenita eseguiti in questo centro sono stati portati a termine con successo in toracoscopia, senza necessità di passare alla tecnica toracotomica né per motivi chirurgici né anestesiologici.
Nel panorama degli interventi chirurgici sul neonato/lattante, la lobectomia polmonare toracoscopica rappresenta un intervento di chirurgia maggiore che prevede la necessità di operare molto vicino al cuore, su vasi sanguigni di grandi dimensioni e sulle principali diramazioni dei bronchi. Dopo aver eseguito la lobectomia, il tessuto polmonare asportato viene fatto uscire dal torace attraverso una delle cicatrici chirurgiche che viene ampliata. Il materiale asportato viene sottoposto ad esame istologico per la conferma diagnostica. Sulla cute si applica la colla che crea un film impermeabile.
Dopo l’intervento chirurgico, il bambino non ha generalmente bisogno di una degenza, pur temporanea, in terapia intensiva. Per le prime 24 ore post-operatorie, il bambino è mantenuto in osservazione in Terapia Intensiva Pediatrica. In entrambi i casi di lobectomia o segmentectomia, nel torace viene posizionato un drenaggio che resterà in sede per 1 o 2 giorni dopo l’intervento. Dopo qualche giorno il drenaggio toracico sarà rimosso (in genere al 3°-5° giorno). Avrà nel torace un piccolo tubo che serve da drenaggio e sarà sottoposto a terapia antidolorifica. Qualche ora dopo l'intervento riprenderà ad alimentarsi per bocca con la sua dieta consueta. È importante sapere che il reparto è aperto ai genitori 24 ore su 24, fatti salvi alcuni momenti particolari. Nel corso di tutto il ricovero, la presenza dei genitori accanto al bambino è molto importante, poiché ha bisogno di essere rassicurato, accudito e coccolato. Standogli accanto e osservando le sue reazioni, la mamma e il papà si renderanno conto dei suoi bisogni e, con l'aiuto del personale, proveranno a soddisfarli.
Talora i quadri di CPAM possono essere misconosciuti sia in età fetale sia post-natale e pertanto questo percorso diagnostico-terapeutico può non essere seguito. Spesso, in tali bambini più grandi, l’insorgenza di infezioni polmonari ricorrenti o il manifestarsi di pneumotorace (raccolta di aria all'interno del torace che limita la ventilazione del polmone stesso) induce a sospettare l’esistenza di una malformazione polmonare congenita anche in assenza di una storia di diagnosi prenatale o di anomalie non riscontrate nel periodo prenatale.
Follow-up a Lungo Termine e Qualità di Vita
Per tutti i bambini operati, dopo l’intervento viene impostato un accurato programma di follow-up clinico e strumentale multidisciplinare a lungo termine per seguirne la crescita e monitorarne la funzionalità respiratoria e la qualità di vita. Dopo la dimissione dall'ospedale, il bambino eseguirà regolari controlli ambulatoriali, inizialmente più ravvicinati nel tempo, fino all'adolescenza, e in Day-Hospital fino all'anno di vita. Si ritiene, infatti, importante garantire a questi bambini un adeguato e duraturo programma di controlli, che permettano di valutare il loro stato di salute e studiare lo sviluppo della gabbia toracica, dei polmoni e della loro funzionalità. In particolare, gli aspetti che maggiormente meritano di essere controllati sono rappresentati dallo sviluppo polmonare e dallo sviluppo muscolo-scheletrico.
Le malformazioni polmonari congenite, se trattate correttamente, non hanno conseguenze significative sulla qualità di vita dei bambini. La qualità di vita del bambino sarà buona. La capacità respiratoria del polmone residuo è tale da non imporre alcuna limitazione per le attività sportive anche di tipo agonistico. Si può affermare che nei bambini operati la qualità della vita risulta essere buona.
Approcci Terapeutici e Prospettive Future
Siamo consapevoli dei dubbi e dei timori che i genitori potrebbero avere di fronte al sospetto di una malformazione polmonare congenita nel proprio figlio, alla successiva conferma diagnostica ed eventuale indicazione chirurgica. È per questo motivo che si ritiene importante fornire informazioni adeguate su questa patologia in modo che i genitori possano comprendere e condividere il percorso diagnostico-terapeutico proposto.
Si potrebbe sentire che in altri centri offrono programmi terapeutici diversi, basati sul follow-up e l’eventuale intervento solo in caso di modificazioni del quadro radiologico o insorgenza di complicanze. Basandosi sulle conoscenze attualmente disponibili sulle CPAM, l'approccio di sorveglianza non è ritenuto sufficientemente sicuro. Avendo sviluppato in centri specializzati, con un'adeguata formazione anche internazionale, le capacità per eseguire in sicurezza l’intervento di asportazione delle CPAM con la tecnica toracoscopica e quindi con un impatto mini-invasivo sui bambini, si ritiene che l’indicazione all’asportazione di queste malformazioni sia, sulla base delle conoscenze attualmente disponibili, la scelta più sicura. Non esiste al momento attuale una dimostrazione certa della relazione tra malformazioni polmonari congenite e neoplasie, ma con la ricerca si sta lavorando per comprendere più a fondo i meccanismi alla base di queste malformazioni con l’obiettivo di arrivare un giorno a capire meglio la loro evoluzione e potere differenziare forme che necessitino dell’intervento chirurgico da forme per cui può essere sufficiente il follow-up.
Un obiettivo è per esempio quello di arrivare a differenziare le forme che hanno un potenziale maligno da quelle benigne, e già qualche passo avanti è stato fatto. Sappiamo infatti che la presenza in un individuo di mutazioni nel gene DICER-1 predispone allo sviluppo di una forma tumorale, il Blastoma Pleuro Polmonare (PPB). La ricerca di mutazioni in questo gene non permette però attualmente una diagnosi certa e non è quindi ancora possibile differenziare con sicurezza assoluta le forme neoplastiche da quelle puramente malformative.
Le CPAM hanno una buona prognosi complessiva, ma la ricerca continua per migliorare ulteriormente la comprensione e la gestione di queste complesse condizioni.