Quando un genitore bacia il proprio neonato sulla fronte e nota un sapore insolitamente salato sulla pelle, il pensiero va raramente a un problema medico. Eppure, questo dettaglio tattile e gustativo apparentemente trascurabile è uno dei segnali più antichi e noti della fibrosi cistica, una malattia genetica rara e complessa. Questa condizione congenita altera radicalmente il modo in cui il corpo gestisce liquidi e secrezioni, rendendole estremamente dense e difficili da smaltire. Nel neonato, questo può provocare difficoltà respiratorie, problemi intestinali e scarso assorbimento dei nutrienti, condizioni che, pur non manifestandosi sempre con agitazione diretta, possono generare un significativo disagio e irrequietezza.
Accogliere un bambino è un momento di grande emozione, ma anche di tante domande. Nei primi giorni di vita ogni genitore impara a conoscere il proprio piccolo e a interpretare ogni segnale, spesso con una naturale preoccupazione per la sua salute. È in questo contesto che una comprensione approfondita della fibrosi cistica diventa cruciale, per individuare precocemente ogni indizio e garantire un intervento tempestivo.
La Fibrosi Cistica: Una Malattia Genetica Multiorgano
La fibrosi cistica, nota anche come mucoviscidosi, è una malattia genetica ereditaria che può manifestarsi fin dalla nascita. È causata da una mutazione nel gene CFTR (Cystic Fibrosis Transmembrane Conductance Regulator). Il suo malfunzionamento provoca una marcata carenza di cloro e acqua all’interno delle secrezioni cellulari. Di conseguenza, fluidi che dovrebbero agire da lubrificanti naturali si trasformano in un muco viscoso e disidratato. Il gene CFTR, quando funziona normalmente, codifica una proteina responsabile della regolazione dell’acqua e degli elettroliti attraverso le membrane cellulari. La conseguenza è la produzione di secrezioni più spesse e viscose nell’apparato respiratorio, in quello digerente e in quello riproduttivo.
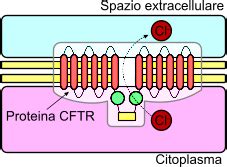
Questa malattia è ereditaria, cioè si eredita al momento del concepimento, quando il bambino eredita i geni malati di origine paterna e materna. La trasmissione avviene solo se entrambi i genitori sono portatori della mutazione del gene CFTR. Il gene malato, ereditato dai genitori, è recessivo; questo significa che i bambini, per ammalarsi, devono ereditare due copie del gene, una per ciascun genitore. I soggetti portatori sani non hanno alcun sintomo della malattia e spesso non sanno nemmeno di esserlo. Se però due portatori sani hanno un figlio, questi ha una probabilità su quattro di ereditare la mutazione da entrambi i genitori, avendo così la malattia conclamata. I soggetti con una copia difettosa sono portatori, ma non hanno la malattia.
La fibrosi cistica è una delle malattie genetiche ereditarie più diffuse nel nostro paese, nonostante sia classificata come rara. In Italia si stima che un neonato ogni 2500-3000 nasca con la malattia. Negli Stati Uniti, colpisce circa 1 neonato caucasico su 3.300 e 1 neonato di colore su 15.300. È rara nelle persone di origine asiatica. Nel mondo, circa il 4% dei soggetti caucasici è portatore di una copia difettosa del gene CFTR. Oggi in Italia vi sono circa 6.000 malati. Questi dati spiegano perché in Italia esista una rete nazionale di centri specializzati e perché lo screening neonatale sia diventato obbligatorio.
Le ricerche mediche e i centri specializzati confermano che questo accumulo anomalo finisce per ostruire progressivamente organi vitali, causando danni particolarmente severi a carico dei bronchi, dei polmoni e dell’apparato digerente. La fibrosi cistica è pertanto una malattia multiorgano. I polmoni sono normali alla nascita, ma successivamente possono presentarsi problemi in qualunque momento quando le secrezioni dense iniziano a ostruire le piccole vie aeree (cosiddetti tappi di muco).
I Segnali nel Lattante: Cosa Osservare Attentamente
I tempi e i modi in cui la malattia si manifesta variano ampiamente. A quest'età potrebbe non esserci ancora alcun sintomo manifesto, pur in presenza di sicura malattia: è ciò che si può verificare per alcuni bambini diagnosticati attraverso screening neonatale. Molti neonati con fibrosi cistica, forse la maggioranza, a un mese di vita non hanno manifestato nessun minimo sintomo di FC.

Tuttavia, prestare attenzione ai segnali fisici prolungati è un passo fondamentale per tutelare la salute.
Il "Bacio Salato": Un Indizio Antico
Un genitore può osservare la formazione di cristalli di sale o perfino un sapore salato sulla cute del bambino. Le ghiandole sudoripare secernono fluidi contenenti più sale del normale, aumentando il rischio di disidratazione. Questo è un sintomo caratteristico dei bambini con fibrosi cistica.
Problemi Digestivi Precoci: Indicatori Cruciali
In genere i sintomi digestivi e nutrizionali sono i primi a manifestarsi. Un segno precoce può essere l’ileo meconiale, un blocco intestinale presente già alla nascita. Si verifica quando il meconio (le feci catramose, di colore nerastro e verdastro che normalmente il neonato produce durante i primissimi uno o due giorni di vita) diventa talmente spesso da ostruire l’intestino. L’ostruzione dell’intestino tenue è definita ileo da meconio e nel colon è definita sindrome da tappo di meconio. Alcuni neonati FC hanno un ritardo di emissione del meconio (che avviene di solito in prima giornata) che giustifica il sospetto. Il 20% circa dei neonati con fibrosi cistica presenta ileo da meconio, che causa vomito, gonfiore (distensione) addominale e assenza di movimenti intestinali. L’ileo meconiale talvolta è complicato dalla perforazione intestinale, una grave condizione che causa infezione e peritonite (infiammazione del tessuto che riveste la cavità e gli organi addominali) e, se non trattata, shock e decesso. I neonati con ileo meconiale quasi sempre sviluppano successivamente altri sintomi della fibrosi cistica.
Spesso il primo sintomo di fibrosi cistica in un neonato che non presenta un ileo meconiale è un ritardo della capacità di riacquistare peso alla nascita, oppure un insufficiente incremento di peso a 4-6 settimane di vita. Questo scarso aumento di peso è dovuto a scarso assorbimento delle sostanze nutritive legato a quantità inadeguate di enzimi pancreatici. Alcuni neonati FC hanno un basso peso alla nascita pur con parto a termine. Può esservi un ritardo di recupero del peso neonatale (che di solito si ha in pochi giorni, dopo qualche giorno di calo fisiologico), come può esservi nel primo mese una crescita in peso stentata (quando c’è insufficienza pancreatica precoce). Spesso il neonato presenta feci frequenti, abbondanti, maleodoranti e oleose (chiamate steatorrea) e può presentare gonfiore (distensione) addominale. Senza trattamento, l’aumento di peso è lento a dispetto di un appetito normale o robusto. Nei bambini, un campanello d’allarme evidente è lo scarso accrescimento in peso e altezza. Talora si ha nel primo mese un certo grado di anemia, accompagnato eventualmente a un modesto edema (gonfiore dei tessuti), sempre nei casi con insufficienza pancreatica precoce. Nelle forme più gravi e protratte di malnutrizione si può avere pallore, cute turgida-edematosa: è la sindrome di anemia, edema, iporoteinemia. Queste forme oggi sono rarissime. I lattanti e i bambini molto piccoli non trattati possono osservare la fuoriuscita parziale del retto attraverso l’orifizio anale, una condizione definita prolasso rettale. I neonati con fibrosi cistica alimentati con latte ipoallergenico o con latte di soia possono sviluppare anemia e gonfiore agli arti, per un malassorbimento proteico.
Il test del portatore sano di fibrosi cistica: per non trasmettere la mutazione ai tuoi figli
Sintomi Respiratori: La Tosse Persistente e le Infezioni
I sintomi respiratori iniziano più o meno precocemente e sono caratterizzati da tosse protratta e insistente, che accompagna o segue, prolungandoli, episodi anche banali di infezione respiratoria. I segnali respiratori da monitorare con attenzione comprendono una tosse persistente, che inizia in modo stizzoso per poi diventare catarrale, e un respiro sibilante accompagnato da una frequente sensazione di affanno. Possono comparire infezioni bronchiali e polmonari frequenti, come bronchiti o polmoniti che faticano a guarire. Talora si ha nel primo mese una tossetta di apparente scarso rilievo (di solito indice di infezione respiratoria precoce). Il facile ricadere di infezioni respiratorie e il loro protrarsi oltre i limiti abituali, in un bambino che non cresce molto, dovrebbe far prendere in considerazione, tra le varie cause possibili, anche la fibrosi cistica.
Disidratazione e Perdita di Sali
Il rischio di disidratazione è un sintomo caratteristico dei bambini con fibrosi cistica quando il clima è caldo umido. Questa condizione, infatti, aumenta la perdita di liquidi e di sali minerali con la sudorazione. I sintomi da perdita di sali possono essere acuti, come disidratazione e collasso, specie in occasione di grandi calure, con forte sudorazione, o di febbre protratta e magari di vomito. Ne è causa l'alta concentrazione di sale nel sudore, che viene perduto con la sudorazione. La perdita di sali può dare anche sintomi cronici, di difficile valutazione e interpretazione, come perdita di appetito, nausea e vomito, astenia, malessere generale.
L'Impatto della Fibrosi Cistica sui Sistemi Vitali
La fibrosi cistica interessa molti organi in tutto l’organismo e quasi tutte le ghiandole che secernono liquidi all’interno di un dotto (ghiandole esocrine).
L'Apparato Respiratorio Sotto Pressione
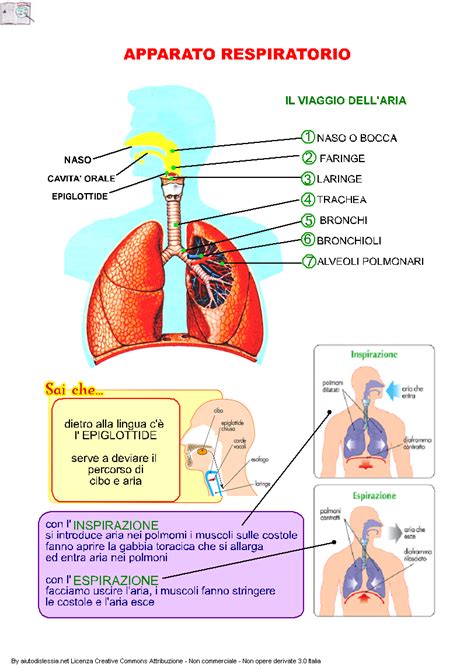
L’apparato respiratorio è solitamente quello più sotto pressione. In questa malattia il muco prodotto nelle vie aeree è molto denso, cosicché la sua normale funzione antibatterica è alterata, favorendo la comparsa di gravi infezioni polmonari. L’ostruzione causata dal ristagno del muco purulento crea l’ambiente ideale per batteri e infiammazioni continue. Con il tempo, compaiono le cosiddette bronchiectasie, dilatazioni dei bronchi dovute al ristagno cronico del muco nelle vie aeree. Questo favorisce la comparsa di infezioni a causa dei batteri che proliferano nel muco e creano uno stato di infiammazione persistente. Questi problemi rendono la respirazione sempre più difficile e riducono la capacità del polmone di trasferire l’ossigeno al sangue. Nei casi più severi si verifica l’emottisi (emissione di sangue dalle vie aeree). Il ripetersi di questi episodi infettivi tende a logorare i tessuti polmonari, portando gradualmente i pazienti verso una progressiva difficoltà respiratoria. Si sviluppano anche frequenti infezioni batteriche del tratto respiratorio che interessano i seni paranasali. La fibrosi cistica è pertanto una malattia multiorgano. Prima di tutto, è una malattia dell’apparato respiratorio e questo, nella maggior parte di casi, influenza la qualità di vita.
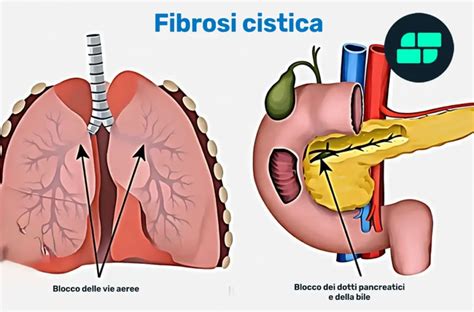
Le Conseguenze sul Sistema Digestivo e Nutrizionale
L’impatto sul sistema digestivo è altrettanto gravoso. L’insufficienza pancreatica colpisce circa l’85% delle persone affette. I dotti che collegano la ghiandola all’intestino si bloccano, impedendo agli enzimi di svolgere il loro lavoro fondamentale per l’assimilazione del cibo. L’ostruzione dei dotti del pancreas impedisce agli enzimi digestivi di raggiungere l’intestino tenue. L’assenza di questi enzimi causa scarso assorbimento dei grassi, delle proteine e delle vitamine (malassorbimento). Questo malassorbimento si traduce in diarrea cronica con feci oleose, difficoltà a digerire i grassi e una grave carenza di vitamine essenziali. Questo scarso assorbimento a sua volta può causare deficit nutrizionali e scarsa crescita. Alla fine, possono formarsi delle cicatrici nel pancreas, che non produce più abbastanza insulina, causando in alcune persone lo sviluppo del diabete. Tuttavia, una piccola percentuale dei pazienti affetti da fibrosi cistica e portatori di certe varianti non sviluppa problemi digestivi a livello del pancreas.
L’intestino può essere ostruito dalle secrezioni dense. I bambini più grandi e gli adulti possono anche avere problemi di stipsi e ostruzione intestinale (detta sindrome da ostruzione intestinale distale). Possono verificarsi disturbi al fegato. La stasi biliare e lo sviluppo del diabete possono essere favoriti negli adulti dall'evoluzione della patologia. L’ostruzione dei dotti biliari dovuta alle secrezioni dense può determinare infiammazione e infine cicatrizzazione del fegato (cirrosi) nel 3-4% circa degli adulti con fibrosi cistica. La cirrosi può aumentare la pressione nelle vene che entrano nel fegato (ipertensione portale), causando una dilatazione e un indebolimento delle vene nella parte inferiore dell’esofago (varici esofagee), che possono rompersi e sanguinare abbondantemente. In quasi tutti i soggetti con fibrosi cistica la cistifellea è piccola, piena di bile densa e malfunzionante. Alcuni soggetti sviluppano calcoli biliari, ma solo una piccola percentuale lamenta sintomi.
Manifestazioni Meno Comuni e Sistematiche
Altri sintomi sistemici includono sinusite cronica, poliposi nasale, stipsi severa e infertilità maschile dovuta ad azoospermia. Nei piccoli di sesso maschile, la fibrosi cistica è caratterizzata dall’ostruzione dei dotti spermatici o atresia dei vasi deferenti. Le secrezioni dense possono bloccare gli organi riproduttivi con conseguente infertilità, soprattutto negli uomini, e molto meno nelle donne. Quasi tutti gli uomini presentano una conta spermatica bassa o assente (che li rende infertili). Nelle donne, le secrezioni della cervice uterina sono troppo dense e riducono la fertilità. Tuttavia, molte donne con fibrosi cistica hanno portato a termine la gravidanza. L’esito della gravidanza per madre e neonato spesso dipende dallo stato di salute della madre durante la gravidanza. D’altro canto, la funzione sessuale maschile o femminile non è interessata.
La fibrosi cistica presenta molte complicanze. L’assorbimento insufficiente delle vitamine liposolubili A, D, E e K determina talvolta cecità notturna, osteopenia (riduzione della densità ossea), osteoporosi, anemia e disturbi emorragici. Altre complicanze possono includere artrite, dolore cronico, problemi del sonno e apnea ostruttiva del sonno, calcoli renali, malattia renale, depressione e ansia, perdita uditiva neurosensoriale e ronzii nell’orecchio (tinnito) causati dall’esposizione a farmaci che danneggiano le orecchie (specialmente gli aminoglicosidi), infezioni croniche dei seni paranasali e un aumento del rischio di tumori dei dotti biliari, del pancreas e dell’intestino. Le malattie polmonari col tempo possono provocare un collasso della parte inferiore destra del cuore (ventricolo destro).
La Diagnosi Precoce: Un Vantaggio Fondamentale
Oggi la maggioranza delle diagnosi avviene nei primissimi giorni di vita grazie allo Screening Neonatale. Oggi la diagnosi di fibrosi cistica nei neonati si basa su una serie di test sicuri e affidabili, che consentono di intervenire prima ancora che i sintomi si manifestino. Ricevere una diagnosi di fibrosi cistica nelle prime settimane di vita può spaventare, ma è in realtà un vantaggio fondamentale. Una diagnosi tempestiva, quindi, non è solo una notizia da affrontare, ma il punto di partenza di un percorso di cura e di fiducia nel futuro. I tempi e i modi in cui la malattia si manifesta variano ampiamente.
Lo Screening Neonatale: Il Primo Passo Obbligatorio
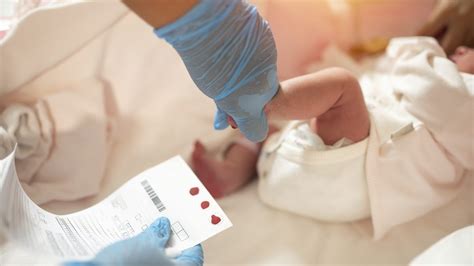
In Italia, lo screening neonatale è obbligatorio e viene eseguito su tutti i neonati nei primi giorni di vita tramite il prelievo di una goccia di sangue dal tallone. Lo screening neonatale consiste in un piccolo prelievo di sangue, effettuato dal tallone del neonato tra le prime 48/72 ore di vita. Si misura il livello di tripsina (un enzima pancreatico che interviene nella digestione) in una goccia di sangue prelevata su carta da filtro. Il primo indicatore ricercato è il tripsinogeno immunoreattivo (IRT), una proteina pancreatica che risulta elevata nei neonati con fibrosi cistica. Se il livello di tripsina nel sangue è alto, alcuni Stati negli Stati Uniti richiedono la ripetizione degli esami. Se dal prelievo ematico emergono valori anomali, si procede con il test del sudore. È importante sottolineare che un risultato positivo allo screening non equivale a una diagnosi, ma segnala solo la necessità di ulteriori accertamenti.
Le Linee Guida internazionali raccomandano l'applicazione dei test diagnostici per la Fibrosi Cistica in tutti i soggetti con sintomi che possono essere legati alla fibrosi cistica, di qualsiasi età o etnia, anche in caso di screening neonatale negativo. Il test di screening neonatale fatto su goccia di sangue raccolta su carta bibula si basa sulla determinazione del livello di Tripsina nel sangue del neonato. Oggi quasi tutti i Centri di Screening procedono subito al test genetico quando il test della tripsina dà risultati oltre la soglia di normalità. I protocolli di screening neonatale per la Fibrosi Cistica prevedono un re-testing (si ripete il dosaggio della tripsina in genere tra 20 e 30 giorni di vita) in caso di positività al primo test effettuato a 3 giorni di vita. Se il retesting risulta normale il procedimento diagnostico si conclude con l’esclusione della malattia. I risultati anormali al 3° giorno di vita costituiscono un falso positivo. Nella maggior parte dei centri di screening, per i casi positivi al primo test viene fatto sulla stessa goccia di sangue anche il test genetico per escludere che siano in causa mutazioni del gene CFTR.
Il Test del Sudore: Il "Gold Standard"
Se dal prelievo ematico emergono valori anomali, si procede con il test del sudore. Questo esame misura in modo indolore la concentrazione di sale prodotta dalle ghiandole sudoripare e rimane lo standard di riferimento clinico per confermare la patologia. Dopo lo screening, il test del sudore è considerato da decenni l’esame di riferimento per confermare la fibrosi cistica. Si tratta di un test non invasivo e indolore, che misura la concentrazione di cloro presente nel sudore. Il test può essere eseguito anche nei primi mesi di vita e fornisce risultati affidabili in poco tempo. Nonostante i progressi tecnologici, il test del sudore rimane il gold standard per la conferma diagnostica. La procedura prevede la stimolazione della sudorazione su una piccola area del braccio (solitamente tramite pilocarpina e una debole corrente elettrica) e la successiva misurazione della concentrazione di cloruro.
Il test del sudore è il modo più efficace per diagnosticare la FC. Il limite al di sopra del quale il test è ritenuto positivo confermando la diagnosi di FC equivale a 60 mEq/L, mentre risulta negativo con valori di cloro al di sotto di 40 mEq/L. Per i valori all’interno di questo range il test può essere ripetuto per dare una diagnosi accertata di FC. Il test viene richiesto nei bambini con screening neonatale positivo per FC, nei bambini che abbiano presentato alla nascita ileo da meconio, per bambini con sintomi respiratori o gastrointestinali che inducano a sospettare una diagnosi di FC e nei casi di pancreatite cronica o grave disidratazione, soprattutto nei mesi estivi.
L'Analisi Genetica: Identificare la Mutazione Specifica
Quando serve maggiore precisione, l’analisi del gene CFTR, che regola il passaggio di cloro e sodio nelle cellule, consente di confermare la diagnosi e di individuare la tipologia di mutazione. La presenza di 2 geni della fibrosi cistica anomali (varianti) è compatibile con la diagnosi di fibrosi cistica. Tuttavia, per confermare la diagnosi è comunque necessario un test del sudore positivo. L’analisi del DNA è fondamentale non solo per confermare la diagnosi, ma anche per identificare la specifica mutazione genetica (ne esistono oltre 2.000). Conoscere la mutazione è oggi indispensabile per stabilire l’eleggibilità ai nuovi farmaci modulatori. Il test genetico è importante anche a fini terapeutici.
Per chi sta pianificando una gravidanza, conoscere il proprio stato genetico è un passo importante. I test genetici pre-concepimento e prenatali consentono di verificare se uno o entrambi i genitori sono portatori della mutazione del gene CFTR. Essere portatore non significa essere malati, ma implica la possibilità di trasmettere la mutazione ai figli. Per chi desidera informarsi prima del concepimento, il test Genescreen® di Eurofins Genoma rappresenta un alleato prezioso. Si tratta di un test genetico pre-concepimento completo e non invasivo, che analizza in un’unica seduta più di 200 malattie ereditarie, tra cui anche la fibrosi cistica. 1 individuo su 25/26 è portatore asintomatico del gene responsabile della FC. Se siete incinta e l’esame evidenzia che vostro figlio potrebbe essere a rischio di fibrosi cistica, il medico potrà consigliare ulteriori esami per il vostro bambino. Va inoltre tenuto presente che alcune mutazioni genetiche della fibrosi cistica non possono essere individuate dall’esame, così com’è concepito attualmente.
Quando la Diagnosi è Meno Chiara: La CRMS
Alcuni bambini con test di screening neonatale positivo per la fibrosi cistica possono essere difficili da classificare, anche dopo il test del sudore o i test genetici. Questi bambini non hanno sintomi correlati alla fibrosi cistica, i risultati dell’analisi del sudore sono al limite della positività e della negatività e non presentano varianti genetiche della fibrosi cistica o ne presentano solo una. I medici diagnosticano questo gruppo come affetto da sindrome metabolica correlata a CFTR (CFTR-related metabolic syndrome, CRMS), chiamata anche diagnosi inconcludente di fibrosi cistica positiva allo screening. Tuttavia esiste un certo numero di falsi negativi allo screening per la Fibrosi Cistica e sono proprio le forme atipiche, con mutazioni del gene CFTR notoriamente legate a forme meno gravi della malattia, che possono essere diagnosticate più tardi. In caso di diagnosi tardiva con screening neonatale negativo il test del sudore ed il test genetico potrebbero non essere conclusivi.
Il test del portatore sano di fibrosi cistica: per non trasmettere la mutazione ai tuoi figli
Le Strategie Terapeutiche Attuali: Dalla Gestione ai Modulatori
Oggi, grazie alla diagnosi precoce e ai trattamenti mirati, l’obiettivo non è più solo convivere con la fibrosi cistica, ma vivere bene, a lungo e con fiducia nel futuro. Oggi la medicina di precisione permette di personalizzare i trattamenti in base al profilo genetico del piccolo paziente, rendendo la gestione della malattia sempre più efficace e “su misura”. Se le cure sono iniziate sin dai primi mesi di vita, possono migliorare la crescita, aiutare a mantenere sani i polmoni, ridurre i ricoveri ospedalieri e prospettare una migliore qualità di vita.
Un Approccio Multidisciplinare e Personalizzato
Il bambino viene preso in cura da un’équipe di diversi esperti presso un centro specializzato. I centri di riferimento regionali garantiscono una presa in carico continua, con controlli periodici e terapie personalizzate. L’équipe di specialisti è in grado di evidenziare i primi segnali o cambiamenti nell’andamento della malattia, intervenendo in maniera appropriata e tempestiva.
Terapie Essenziali per la Gestione Quotidiana
La fisioterapia respiratoria quotidiana serve a mantenere le vie aeree libere dal muco, prevenendo le infezioni respiratorie. Il precoce inizio di una specifica fisioterapia respiratoria è fondamentale per migliorare la prognosi nel tempo. Poiché la malattia può compromettere il funzionamento del pancreas, vengono prescritti enzimi digestivi sotto forma di capsule o polveri da somministrare con il pasto. La quantità da somministrare deve essere valutata tenendo conto della gravità della compromissione pancreatica e delle abitudini alimentari dei pazienti, che devono sottoporsi ad una dieta libera, equilibrata, ipercalorica.
Il muco vischioso che ostruisce le vie respiratorie favorisce la proliferazione di batteri. L’antibioticoterapia (meglio se per via aerosolica) ha un ruolo importante nel controllo delle infezioni. Questa dovrebbe essere prescritta solo in base al risultato dell’esame microbiologico sulle secrezioni bronchiali ottenute dopo la tosse o mediante aspirato faringeo.
La Rivoluzione dei Farmaci Modulatori CFTR
Negli ultimi anni, l’introduzione dei farmaci modulatori CFTR ha rappresentato una vera rivoluzione. Questi trattamenti agiscono direttamente sul difetto genetico, migliorando la funzione della proteina CFTR e riducendo in modo significativo i sintomi respiratori e digestivi. Questi farmaci molecolari (come l’associazione elexacaftor/tezacaftor/ivacaftor) agiscono direttamente alla base del difetto biologico, aiutando la proteina CFTR a posizionarsi correttamente sulla membrana cellulare e a funzionare meglio. Oggi, accanto alle terapie di routine, per alcune mutazioni come ad esempio la F508del e ed altre meno frequenti, sono disponibili farmaci correttori e potenziatori che intervengono sul funzionamento della proteina CFTR, migliorando il decorso della malattia.
I modulatori genici della proteina Cftr compensano il difetto genetico alla base della malattia, con risultati spesso molto positivi. Uno recentissimo è basato sui principi attivi deutivacaftor/tezacaftor/vanzacaftor ed è stato approvato da poco dal Comitato per i Medicinali per Uso Umano dell’Agenzia Europea per i Medicinali per il trattamento della fibrosi cistica (FC) in malati di età pari e superiore a 6 anni che presentano almeno una particolare mutazione nel gene Cftr.
Opzioni Avanzate: Il Trapianto e la Terapia Genica Futura
Quando la malattia polmonare raggiunge uno stadio terminale e le terapie mediche non sono più sufficienti, il trapianto bipolmonare rappresenta un’opzione salvavita. Per alcune persone può essere necessario un trapianto di fegato e polmoni.
La terapia genica consiste nel somministrare al malato il gene Cftr attraverso sofisticate tecniche, per esempio per via inalatoria, virus completamente innocui che agiscono da vettori. Questi trattamenti non sono ancora disponibili per tutti, poiché sono indicati solo per alcune mutazioni. In futuro sarà possibile allargare ulteriormente la percentuale di persone che potranno beneficiarne. Inoltre oggi, in caso di diagnosi genetica che non ha un risultato definitivo, si aprono nuove possibilità diagnostiche attraverso studi funzionali della proteina che il gene alterato produce.
Vivere con la Fibrosi Cistica: Aspettative e Qualità della Vita
Fino a pochi decenni fa, la fibrosi cistica era considerata una malattia a prognosi severa. Oggi, grazie alla diagnosi precoce e ai trattamenti mirati, l’obiettivo non è più solo convivere con la fibrosi cistica, ma vivere bene, a lungo e con fiducia nel futuro. L’aspettativa di vita per le persone con fibrosi cistica è notevolmente migliorata negli ultimi decenni grazie ai progressi nella diagnosi precoce e nelle terapie molecolari. Grazie ai miglioramenti delle terapie, che hanno prolungato l’aspettativa di vita dei malati di fibrosi cistica, negli Stati Uniti circa il 60% è costituito da adulti.
Accanto ai progressi farmacologici, si stanno diffondendo programmi di monitoraggio domiciliare e strumenti digitali che permettono alle famiglie di seguire in tempo reale l’andamento della malattia. La chiave è la presa in carico continua: controlli regolari, monitoraggi respiratori e supporto nutrizionale permettono di intervenire tempestivamente su ogni variazione clinica.
L’attività fisica regolare è considerata a tutti gli effetti parte della terapia: lo sport aiuta la clearance del muco, migliora la capacità aerobica e ha effetti positivi sull’umore e sulla densità ossea. È anche importante il supporto psicologico per la gestione quotidiana della malattia.