Nei globuli rossi del feto è possibile identificare una forma di emoglobina diversa da quella adulta. L’emoglobina è contenuta all’interno dei globuli rossi, e il suo ruolo consiste nel trasportare l’ossigeno ai tessuti del nostro organismo. Questa proteina fondamentale è la principale frazione emoglobinica durante la vita fetale e alla nascita costituisce circa l’80% dell’emoglobina totale. La presenza di questa emoglobina speciale è cruciale per la sopravvivenza e lo sviluppo del feto nell'ambiente uterino, dove le condizioni di ossigeno sono significativamente diverse da quelle esterne.
Struttura e Funzione dell'Emoglobina Fetale: Un Adattamento Essenziale
L'emoglobina fetale, conosciuta anche come HbF od emoglobina F, presenta una struttura molecolare specifica che la distingue dall'emoglobina dell'adulto. L'emoglobina dell'adulto è in massima parte HbA ed è costituita da due coppie di catene, denominate α (alfa) e ß (beta). Invece, nell’emoglobina fetale si trovano due catene α e due catene γ (gamma). In particolare, l'emoglobina fetale è formata da due catene α e da due catene γ, costituite rispettivamente da 141 e 146 amminoacidi. Le due catene alfa sono identiche a quelle presenti nell'emoglobina adulta, mentre quelle gamma differiscono dalle Beta per 39 amminoacidi.
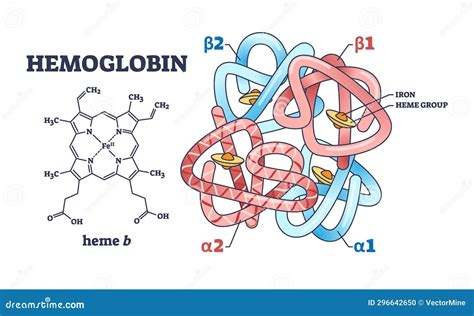
Questa modifica strutturale conferisce all'emoglobina fetale un'affinità per l'ossigeno superiore. In altre parole, si lega all'ossigeno in modo più tenace rispetto all'emoglobina adulta. Dal punto di vista funzionale, questa maggiore affinità permette al feto di estrarre con maggiore efficacia l'ossigeno dal sangue materno. L'emoglobina fetale lega l'ossigeno più fortemente dell'emoglobina materna. Riesce infatti a trasportare percentuali comprese tra il 20% e il 30% di ossigeno in più, rispetto all’emoglobina materna. Il trasferimento di ossigeno al sangue fetale attraverso la barriera placentare è favorito anche dalla maggiore concentrazione di emoglobina, più alta di circa il 50% rispetto a quella del sangue materno. In questo processo l'emoglobina fetale svolge un ruolo essenziale, garantendo che l'ossigeno che attraversa la placenta venga raccolto dall'emoglobina fetale.
Un fattore chiave che contribuisce a questa maggiore affinità è la differente interazione con il 2,3-bisfosfoglicerato (2,3-BPG). L'emoglobina adulta possiede amminoacidi basici che legano il 2,3-BPG, una molecola che riduce l'affinità dell'emoglobina per l'ossigeno. Le catene gamma dell'emoglobina fetale hanno amminoacidi basici differenti e così impediscono la corretta interazione con il 2,3-BPG, assicurando che l'ossigeno si leghi meglio all'emoglobina fetale. Ciò facilita il trasferimento di ossigeno dall'emoglobina adulta della madre a quella fetale. Oltre all'HbF, nelle prime settimane dal concepimento vengono prodotte anche le emoglobine embrionali, come la Portland (ζ2γ2). Ogni molecola di emoglobina è costituita da quattro proteine globulari (catene) strettamente associate tra di loro: ognuna di esse ospita una molecola non proteica, l'eme (un tetrapirrolo a cui è associato un atomo di Ferro, che lega a sé l'ossigeno prelevato dagli alveoli polmonari).
Espressione e Sviluppo: La Transizione dall'Emoglobina Fetale all'Adulto
L'espressione nel tempo e nei diversi tessuti dei differenti tipi di catene globiniche umane è un processo finemente regolato. La differente espressione nel tempo, dal concepimento alla vita adulta, delle diverse catene globiniche nell'uomo dipende dall'attivazione e dallo spegnimento di specifici geni. Questo significa che, nel corso dello sviluppo, il corpo umano produce diverse varianti di emoglobina per adattarsi alle mutevoli esigenze fisiologiche. L’emoglobina fetale (HbF) rappresenta la principale frazione emoglobinica durante la vita fetale.
Dopo la nascita, la sintesi di HbF diminuisce gradualmente e viene sostituita da quella dell’emoglobina A (HbA). La sintesi delle globine Beta, caratterizzanti l'emoglobina adulta, appena percettibile durante la vita fetale, raggiunge il normale regime soltanto verso la fine del terzo mese della vita extrauterina. Entro il primo anno di vita, le concentrazioni di emoglobina fetale scendono a livelli generalmente inferiori all'1%. Negli adulti normali, i valori di emoglobina fetale sono compresi tra lo 0.3% e l'1.2%. Oltre all'HbA, in bassa percentuale, si trova l'HbA2, costituita da due catene α e due catene δ (delta), rappresentando meno del 3.5%. La rimanente percentuale (in genere > 96%) è coperta dall'emoglobina di tipo A. L'emoglobina fetale (HbF), costituita da due catene α e due catene γ (gamma), è l'emoglobina predominante nella vita fetale e diminuisce alle percentuali dell'adulto dopo i primi sei mesi di vita.
LE ANEMIE 💊 H3 Pills
Significato Patologico e Persistenza Ereditaria dell'Emoglobina Fetale (HPFH)
Una piccola percentuale di emoglobina fetale viene espressa anche durante la vita adulta. I suoi livelli possono variare anche di molto sotto l'influenza di fattori quali l'età, il sesso o peculiarità genomiche. Quando negli adulti si hanno valori superiori a 1.1, si parla di emoglobina F alta. In un adulto, livelli di emoglobina fetale considerabili normali oscillano tra 0.1 e 1.1. I valori di riferimento degli esami di laboratorio possono variare a seconda della metodologia di analisi dei campioni; quelli indicati in questa scheda hanno uno scopo puramente informativo. Una precisazione è comunque necessaria per interpretare questi valori. Un'elevata HbF può indicare una condizione di recupero dovuta a ipoplasia di midollo osseo, ovvero un disturbo delle cellule staminali ematopoietiche.
Alcuni soggetti sono affetti dalla cosiddetta persistenza ereditaria dell'emoglobina fetale (HPFH), una condizione benigna in cui concentrazioni importanti di emoglobina fetale (> 10%) persistono anche in età adulta. Si è notato come tale peculiarità, generalmente asintomatica, possa alleviare la severità di certe emoglobinopatie e talassemie. La persistenza ereditaria dell'emoglobina fetale (HPFH) associata a beta talassemia (BT) è caratterizzata da livelli elevati di emoglobina fetale (HbF) e da un aumento del numero delle cellule contenenti l'HbF. L'associazione tra la HPFH e la BT attenua i sintomi clinici nei pazienti, che possono essere asintomatici o presentare una BT intermedia.
La HPFH è dovuta a delezioni nel cluster genico della beta-globina o a mutazioni puntiformi nei geni HBG1 e HBG2 (11p15.5). La diagnosi si basa sulla presenza di un significativo aumento dell'HbF, che varia tra 10 e 40% negli eterozigoti, con indici eritrocitari normali o quasi. L'HbF si distribuisce in maniera omogenea negli eritrociti e i livelli dell'HbA2 sono normali o ridotti. La differenziazione tra la HPFH e la delta-beta talassemia è difficile e può essere confermata dal rapporto di sintesi tra le globine alfa-beta e dall'esame del DNA; non sempre questa differenziazione è possibile sulla base dell'analisi standard del sangue. La trasmissione della HPFH è co-dominante.
Talassemie: Implicazioni della Ridotta Produzione di Emoglobina
Le talassemie sono un gruppo di anemie ereditarie, caratterizzate da un difetto quantitativo nella sintesi dell'emoglobina (Hb). Alla base di tali disordini, vi sono alterazioni genetiche eterogenee che determinano l'abolizione o la riduzione della produzione di una o più catene globiniche. Ne consegue una riduzione nella sintesi dell'emoglobina, che conduce ad un quadro di anemia e di eritropoiesi inefficace di gravità variabile. Le talassemie sono causate da un difetto ereditario che impedisce la normale sintesi delle catene α (α-talassemia) o delle catene ß (ß-talassemia).
Sulla base della catena mancante, si distinguono α-talassemie e β-talassemie. L'informazione genetica per le catene α è contenuta in 4 geni: nel caso dell'α-talassemia perciò il difetto può derivare dall'alterazione di tutte e quattro i geni, una condizione non compatibile con lo sviluppo del feto. Il difetto può anche derivare dall'alterazione di tre geni, che porta alla presenza di Hb formata da complessi di quattro catene ß chiamata HbH, accompagnata da anemia emolitica, splenomegalia e altri sintomi. L'alterazione di due geni configura il tratto talassemico con presenza di anemia e globuli rossi di volume ridotto, ma con percentuali di HbA2 e HbF di solito normali. Infine, l'alterazione di un solo gene è una condizione indistinguibile dalla normalità, ma con possibile trasmissione del carattere alla prole.
L'informazione genetica per le catene ß è contenuta in 2 geni: nel caso della ß-talassemia il difetto può derivare dall'alterazione di entrambi i geni, definendo la forma omozigote. In questo caso la malattia è definita ß-talassemia major o morbo di Cooley, che si manifesta subito dopo la nascita con una anemia molto grave, che necessita di trasfusioni periodiche di sangue. Oppure, il difetto può derivare dall'alterazione di un solo gene, e in questo caso viene chiamato ß-talassemia minor o tratto talassemico. La maggior parte delle persone con talassemia minor non presenta sintomi; i globuli rossi nel loro sangue sono in numero maggiore che nel normale, ma di volume ridotto - da cui il termine di microcitemia - e poveri di emoglobina. La riduzione delle catene polipeptidiche β e δ, invece, configura il quadro della ß-δ talassemia. Inoltre, è possibile la persistenza ereditaria dell'emoglobina fetale (Hb F). Le caratteristiche cliniche delle talassemie dipendono dallo specifico difetto genetico.
Emoglobinopatie e Anemia Falciforme: Il Ruolo Protetttivo dell'HbF
Per definizione le emoglobinopatie sono varianti emoglobiniche e vengono ricomprese nei disordini genetici. Attualmente sono stati rilevati circa 1000 tipi di emoglobinopatie. Un esempio significativo è l'anemia falciforme, una malattia del sangue in cui i globuli rossi si piegano a forma di C o di falce. L'emoglobina fetale non è influenzata da questa mutazione che caratterizza l'anemia falciforme. Per questo motivo, i bambini affetti da anemia falciforme, che presentano livelli elevati di emoglobina fetale, manifestano la malattia in forma molto lieve. La presenza di emoglobina fetale nel sangue può ridurre i sintomi della malattia in persone affette da anemia falciforme. Una terapia farmacologica capace di aumentare la concentrazione di emoglobina fetale apporta benefici significativi ad alcune categorie di pazienti, come quelli affetti da anemia falciforme o da talassemia Beta.
Diagnosi e Monitoraggio dei Livelli di Emoglobina Fetale
La misura della HbF è utile nello studio e nella diagnosi di alcuni importanti disordini a carico dei geni globinici, dove le concentrazioni di HbF possono aumentare considerevolmente. L'esame per l’emoglobina fetale viene svolto per la diagnosi di patologie a carico del sangue, quali talassemia e anemia falciforme.Per l’esame dell’emoglobina fetale viene richiesto un digiuno di almeno otto ore. In un adulto, livelli di emoglobina fetale considerabili normali oscillano tra 0.1 e 1.1. Quando negli adulti si hanno valori superiori a 1.1, si parla di emoglobina F alta.

Nel contesto della gravidanza, la diagnosi prenatale è cruciale. Per rilevare durante la gravidanza se un feto è affetto da talassemia maior, è possibile determinare la quantità di emoglobina adulta presente in un campione di sangue prelevato mediante cordocentesi. Nell'utero, il feto normale produce una piccola quota di emoglobina adulta (2,5-5%). Il feto con talassemia maior ne produce ancor meno (inferiore al 2%). Questo consente una diagnosi precoce e la pianificazione di interventi.
Si parla di “anemia” quando la concentrazione di emoglobina o dell’ematocrito è inferiore all’atteso per i valori noti in epoca neonatale. Nella vita in utero, l’emoglobina fetale (HbF) ha una maggior affinità per l’ossigeno (e quindi minor rilascio nei tessuti) rispetto all’emoglobina adulta. Per questo motivo sono stati individuati dei segni ecografici indiretti di anemia fetale. L’idrope fetale, ad esempio, si identifica con la presenza di raccolte di liquido in almeno due diversi distretti corporei. La valutazione della velocimetria Doppler della MCA (Arteria Cerebrale Media) può indicare anemia fetale anche in casi di alloimmunizzazione anti Kell, nonostante, in questi casi, l’anemia non sia dovuta solo ad emolisi ma anche ad una soppressione dell’eritropoiesi midollare. La valutazione della PSV-MCA può diagnosticare anemia fetale dovuta anche ad infezione da parvovirus, Citomegalovirus, o anemia secondaria ad emorragia feto-materna. Nel caso di sospetto forte di anemia fetale, soprattutto se ci sono già segni di idrope, è indicato il trattamento con la trasfusione di sangue in utero.
La trasfusione di sangue in utero è una procedura invasiva, che prevede l’inserzione di un ago attraverso l’addome materno, fino a raggiungere l’addome del feto, dove si trova l’ingresso della vena ombelicale, o il cordone ombelicale nel punto in cui si inserisce sulla placenta. Prima della procedura il feto viene sedato mediante l’iniezione intramuscolare di una piccola quantità di sostanza (curaro) necessaria per ridurre i movimenti fetali. Si procede quindi al prelievo di una piccola quantità di sangue (dal cordone ombelicale o dalla porzione intraepatica della vena ombelicale) per verificare l’entità dell’anemia e calcolare la quantità di sangue fetale che deve essere trasfusa. Tutto avviene sotto controllo ecografico continuo.
Approcci Terapeutici e Modulazione Farmacologica dell'Emoglobina Fetale
La modulazione dei livelli di emoglobina fetale rappresenta una strategia terapeutica promettente per diverse patologie ematologiche. Una terapia farmacologica capace di aumentare la concentrazione di emoglobina fetale apporta benefici significativi ad alcune categorie di pazienti, come quelli affetti da anemia falciforme o da talassemia Beta. Il prototipo di questi farmaci è stata l'idrossiurea, un farmaco antineoplastico ad azione mielosopressiva. L'idrossiurea si è dimostrata efficace nell'aumentare i livelli di emoglobina fetale e nel ridurre l'incidenza di crisi dolorose in pazienti affetti da anemia falciforme. La sua azione nel promuovere la produzione di HbF è fondamentale per alleviare la gravità della malattia.
Studi recenti hanno anche identificato il ruolo di geni specifici, come BCL11A, nella regolazione della sintesi di emoglobina fetale. La manipolazione di questi geni potrebbe offrire nuove vie per terapie future volte ad aumentare i livelli di HbF. L'aumento dell'emoglobina fetale, indotto farmacologicamente o attraverso altre metodologie, può quindi mitigare gli effetti deleteri delle emoglobinopatie, migliorando la qualità di vita dei pazienti.
tags: #emoglobina #fetale #iatrogena