Introduzione: L'Emoglobina e il suo Ruolo Vitale
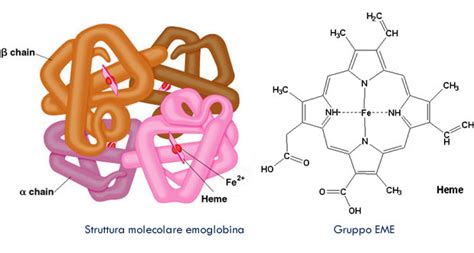
L'emoglobina (Hb) è una proteina essenziale presente nei globuli rossi, il cui compito principale è quello di trasportare l'ossigeno dai polmoni a tutte le parti del corpo e di riportare l'anidride carbonica dai tessuti ai polmoni per l'espirazione. Questa molecola fondamentale è contenuta all’interno dei globuli rossi e rappresenta il veicolo primario per l'ossigeno nel nostro organismo. Ogni molecola di emoglobina è costituita da quattro proteine globulari, note come catene globiniche o globine, strettamente associate tra di loro. Ciascuna di queste subunità presenta al suo interno una struttura chiamata gruppo eme, una molecola non proteica un tetrapirrolo a cui è associato un atomo di ferro. È proprio questo atomo di ferro che lega a sé l'ossigeno prelevato dagli alveoli polmonari, rendendo possibile il trasporto vitale attraverso il flusso sanguigno.
L'efficacia e la stabilità dell'emoglobina sono cruciali per la salute generale dell'individuo. La sua capacità di legare e rilasciare ossigeno in modo efficiente è regolata da complesse interazioni molecolari e strutturali. Un'alterazione, anche minima, in questa complessa architettura può avere ripercussioni significative sulla funzionalità complessiva della molecola e, di conseguenza, sulla capacità del sangue di fornire ossigeno ai tessuti, portando a condizioni patologiche che possono variare da lievi a estremamente gravi. Lo studio di queste alterazioni, note come emoglobinopatie, rientra pienamente nelle scienze biomediche, poiché richiede una comprensione approfondita della genetica, della biologia molecolare, della fisiologia e della patologia per la diagnosi, la gestione e il trattamento.
Le Varie Forme di Emoglobina: Un Panorama Molecolare
Esistono diverse varianti di emoglobina, ognuna con una specifica composizione e funzione, che si alternano e si evolvono nel corso della vita di un individuo, dal concepimento all'età adulta. Fondamentalmente, l'emoglobina è formata da quattro subunità, o catene globiniche, designate come: α (alfa), β (beta), γ (gamma), δ (delta). La combinazione di queste catene determina i diversi tipi di emoglobina presenti nel corpo umano.
Le tipologie principali di emoglobina includono:
- Emoglobina A (HbA): È l'emoglobina predominante dell'adulto, costituita da due catene alfa (α) e due catene beta (ß) (α2ß2). In un adulto sano, l'HbA copre in genere oltre il 96% dell'emoglobina totale.
- Emoglobina A2 (HbA2): Costituisce circa il 2-3.5% dell'emoglobina totale ed è costituita da due catene α e due catene δ (delta) (α2δ2). Un aumento dell'emoglobina A2 è uno dei marker più tipici del portatore di geni ß-talassemici, rendendola un indicatore diagnostico prezioso.
- Emoglobina F (HbF): Conosciuta come emoglobina fetale, rappresenta l’emoglobina predominante nella vita fetale. Negli adulti e nei bambini di età superiore a un anno, l'emoglobina F normalmente non è più dell'1% dell'emoglobina totale, con valori compresi tra lo 0.3% e l'1.2% in adulti normali. È costituita da due catene alfa e due catene gamma (α2γ2).

L'espressione nel tempo, dal concepimento alla vita adulta, delle diverse catene globiniche nell'uomo dipende dall'attivazione e dallo spegnimento di specifici geni. Questo processo di "switch" è cruciale per l'adattamento dell'organismo alle diverse esigenze di ossigenazione nelle varie fasi della vita. Ad esempio, nelle prime settimane dal concepimento, vengono prodotte emoglobine embrionali come l'Emoglobina Portland (ζ2γ2), che vengono poi sostituite dall'emoglobina fetale e, successivamente, dall'emoglobina adulta.
L'Emoglobina Fetale (HbF): Un Adattamento Cruciale per la Vita Pre-Natale
L'emoglobina fetale, o HbF, è la prima emoglobina prodotta durante la vita fetale e riveste un ruolo essenziale nello sviluppo pre-natale. Nei globuli rossi del feto è possibile identificare questa forma di emoglobina, diversa da quella adulta. In particolare, l'emoglobina fetale è formata da due catene α e da due catene γ, costituite rispettivamente da 141 e 146 amminoacidi. Le due catene alfa sono identiche a quelle presenti nell'emoglobina adulta, mentre quelle gamma differiscono dalle beta per 39 amminoacidi.
Questa modifica strutturale conferisce all'emoglobina fetale un'affinità per l'ossigeno superiore; in altre parole, si lega all'ossigeno in modo più tenace rispetto all'emoglobina adulta. Dal punto di vista funzionale, l'emoglobina fetale (HbF od emoglobina F) permette al feto di estrarre con maggiore efficacia l'ossigeno dal sangue materno. Il trasferimento di ossigeno al sangue fetale attraverso la barriera placentare è facilitato non solo da questa maggiore affinità ma anche dalla maggiore concentrazione di emoglobina nel sangue fetale, più alta di circa il 50% rispetto a quella del sangue materno.
La ragione molecolare di questa maggiore affinità è intrinsecamente legata all'interazione con il 2,3-bisfosfoglicerato (2,3-BPG). Nelle catene beta adulte, i gruppi amminoacidici basici positivi legano il 2,3-BPG, una molecola che si lega all'emoglobina e ne riduce l'affinità per l'ossigeno, facilitando il rilascio di ossigeno ai tessuti. Al contrario, le catene gamma dell'emoglobina fetale hanno meno amminoacidi basici in questa regione critica, e così impediscono la corretta interazione con il 2,3-BPG. Questo fa sì che l'emoglobina fetale leghi l'ossigeno più fortemente dell'emoglobina materna, la quale è più influenzata dal 2,3-BPG. Tale differenza favorisce in modo significativo il passaggio di ossigeno dall'emoglobina adulta della madre a quella fetale attraverso la placenta, un processo vitale per la crescita e lo sviluppo del feto.
Normalmente, la produzione di HbF diminuisce progressivamente alla nascita, un processo chiamato "switch emoglobinico". Entro il primo anno di vita, le concentrazioni di emoglobina fetale scendono a livelli generalmente inferiori all'1%. La sintesi delle globine Beta, caratterizzanti l'emoglobina adulta, appena percettibile durante la vita fetale, raggiunge il normale regime soltanto verso la fine del terzo mese della vita extrauterina e si stabilizza su valori molto bassi intorno a 1-2 anni. Adulti normali presentano valori di emoglobina fetale compresi tra lo 0.3% e l'1.2%, meno del 3.5% di emoglobina A2 (α2, δ2), e la rimanente percentuale (in genere > 96%) coperta dall'emoglobina di tipo A.
Emoglobinopatie: Disordini Ereditari della Catena Globinica
Un'emoglobinopatia è un disordine ematico ereditario caratterizzato dalla presenza di forme anomale dell’emoglobina, note come varianti emoglobiniche, o dalla riduzione della produzione della stessa, una condizione conosciuta come talassemia. Queste patologie rientrano nei disordini genetici e attualmente sono stati rilevati circa 1000 tipi di emoglobinopatie.
La causa fondamentale di queste condizioni risiede nella presenza di mutazioni nei geni codificanti per le catene globiniche. Tali mutazioni possono determinare la produzione di catene globiniche strutturalmente alterate, che compongono emoglobine anomale, oppure possono comportare la perdita della produzione di uno o più tipi di catene globiniche, come nel caso delle talassemie. Le mutazioni possono influenzare la struttura dell'emoglobina, la sua funzionalità, la sua velocità di produzione o anche la sua stabilità, con conseguenze variabili sulla salute dell'individuo.
Circa il 7% della popolazione mondiale è portatore in eterozigosi di una variante genetica in una delle catene dell'emoglobina. È importante notare che il tasso di mutazione può variare notevolmente in base all'etnia, indicando una distribuzione geografica e demografica disomogenea di queste condizioni. Per esempio, alcune varianti sono più comuni nelle popolazioni di origine africana o del sud-est asiatico, spesso a causa di un vantaggio evolutivo che i portatori di queste mutazioni hanno acquisito in determinate aree, come la protezione contro la malaria.
È anche possibile che la stessa persona erediti due differenti geni anomali, uno da ciascun genitore, con conseguente combinazione delle varianti emoglobiniche rilevate dai test. Questa condizione è nota come eterozigosi composta o doppia eterozigosi, e può portare a manifestazioni cliniche più complesse o a combinazioni uniche di emoglobinopatie. La comprensione di queste basi genetiche è essenziale per la diagnosi e la consulenza genetica.
Le Talassemie: Alterazioni nella Produzione delle Catene Globiniche
Le Talassemie sono una classe di patologie caratterizzate dalla diminuita produzione o carenza di una delle catene globiniche. Sono causate da un difetto ereditario che impedisce la normale sintesi delle catene α (α-talassemia) o delle catene ß (ß-talassemia). Il nome "talassemia" deriva dal greco "thalassa", che significa mare, in riferimento alla loro elevata prevalenza nelle popolazioni delle regioni costiere del Mediterraneo, ma anche del Medio Oriente, dell'Asia meridionale e del Sud-Est asiatico.
Alfa-Talassemia
L'informazione genetica per le catene α è contenuta in 4 geni, situati sul cromosoma 16. Nel caso dell'α-talassemia, il difetto può derivare dall'alterazione di uno, due, tre o tutti e quattro questi geni, determinando diversi gradi di gravità:
- Alterazione di un gene: Questa condizione è spesso indistinguibile dalla normalità, non presentando sintomi evidenti, ma il portatore può trasmettere il carattere alla prole.
- Alterazione di due geni: Porta al tratto talassemico (o alfa-talassemia minor), caratterizzato da una lieve anemia e globuli rossi di volume ridotto (microcitemia). Di solito, le percentuali di HbA2 e HbF rimangono normali.
- Alterazione di tre geni: Questa condizione comporta la presenza di una forma di emoglobina anomala chiamata Emoglobina H (HbH), composta da quattro catene beta globiniche (ß4). L'HbH viene prodotta in caso di grave carenza delle catene alfa, ed è accompagnata da anemia emolitica, splenomegalia (ingrossamento della milza) e altri sintomi clinici.
- Alterazione di tutti e quattro i geni: Questa è una condizione non compatibile con lo sviluppo del feto (idrope fetale) e spesso porta alla morte in utero o subito dopo la nascita. In questi feti si sviluppa l'Emoglobina di Bart, composta da quattro catene globiniche gamma (γ4), prodotta in caso di grave carenza delle catene alfa in maniera analoga a quanto avviene per l'HbH.
Beta-Talassemia
L'informazione genetica per le catene ß è contenuta in 2 geni, situati sul cromosoma 11. Nel caso della ß-talassemia, il difetto può derivare dall'alterazione di uno o di entrambi i geni:
- Alterazione di un solo gene (forma eterozigote): Questo difetto viene chiamato ß-talassemia minor o tratto talassemico. La maggior parte delle persone con talassemia minor non presenta sintomi significativi, sebbene possano avere un numero maggiore di globuli rossi rispetto alla normalità, ma di volume ridotto (microcitemia) e poveri di emoglobina. L'emoglobina A2 può essere aumentata e l'HbF normale o leggermente aumentata.
- Alterazione di entrambi i geni (forma omozigote): In questo caso, la malattia è definita ß-talassemia major o morbo di Cooley. Si manifesta subito dopo la nascita con una anemia molto grave, che necessita di trasfusioni periodiche di sangue per la sopravvivenza del paziente. I feti con talassemia major producono ancor meno emoglobina adulta (inferiore al 2%) rispetto al feto normale (2,5-5%).
La persistenza ereditaria dell'emoglobina fetale (HPFH) associata a beta talassemia è caratterizzata da livelli elevati di emoglobina fetale (HbF) e da un aumento del numero delle cellule contenenti l'HbF. Questa associazione attenua i sintomi clinici nei pazienti, che possono essere asintomatici o presentare una ß-talassemia intermedia.
Varianti Emoglobiniche Specifiche: Oltre la Normalità
Oltre alle talassemie, esistono molteplici varianti emoglobiniche, alcune delle quali sono silenti, ossia senza segni e sintomi evidenti, mentre altre sono in grado di influenzare significativamente la funzionalità e/o la stabilità della molecola emoglobinica, portando a patologie distintive. Spesso, le forme meno comuni prendono il nome dal luogo di appartenenza della/e famiglia/e in cui la variante genetica è stata identificata per la prima volta.
Emoglobina S (HbS): L'Anemia Falciforme
L'Emoglobina S (HbS) è la variante responsabile dell'anemia falciforme, una delle emoglobinopatie più studiate e clinicamente significative. Si tratta di una patologia a frequenza molto variabile, ma comunque maggiormente presente nelle aree nelle quali la malaria è o è stata endemica. Questa elevata prevalenza è dovuta a un vantaggio evolutivo dei portatori eterozigoti (quelli con "tratto" falciforme) nei confronti della malaria, che li rende più resistenti all'infezione parassitaria.
Le persone affette da anemia falciforme presentano entrambe le copie del gene responsabile della produzione di HbS (stato di omozigosi), e pertanto presentano elevate quantità di HbS. Le persone con "tratto" falciforme (eterozigoti) presentano circa il 40% di HbS e il 60% di HbA normale. La presenza di HbS determina, in condizioni di basse concentrazioni di ossigeno (come può accadere durante un esercizio fisico intenso, in caso di infezioni polmonari, o in alta quota), una variazione della forma dei globuli rossi. Questi assumono la caratteristica forma a falce o a "C", da cui il nome della malattia.
ANEMIA FALCIFORME in 60 secondi o meno - Spiegazione
Gli eritrociti falciformi sono rigidi e possono bloccare i piccoli vasi sanguigni, causando episodi dolorosi acuti, noti come crisi vaso-occlusive, scompensi circolatori e una diminuzione dell'apporto di ossigeno ai tessuti. Questa ridotta ossigenazione, o ipossia, porta a una diminuzione della sopravvivenza cellulare e a danni agli organi. La presenza di emoglobina fetale può alleviare la severità dell'anemia falciforme. È stato notato che i pazienti con anemia falciforme che mantengono elevati livelli di emoglobina fetale manifestano la malattia in forma molto lieve, poiché l'HbF non polimerizza con l'HbS e aiuta a prevenire la falcizzazione.
Emoglobina C (HbC)
L'Emoglobina C (HbC) è un'altra variante beta-globinica. Lo stato di portatore di HbC (eterozigoti) è presente in circa il 2-3% delle persone di origine africana. Lo stato di omozigosi per HbC, pur essendo un evento raro, è responsabile di effetti relativamente lievi, spesso associati a una lieve anemia emolitica e talvolta a splenomegalia.
Emoglobina E (HbE)
L'Emoglobina E (HbE) è una delle varianti beta-globiniche più comuni nel mondo, con una maggiore prevalenza nelle persone originarie del sud-est asiatico. I soggetti omozigoti per HbE in genere hanno una lieve anemia emolitica, macrocitosi (globuli rossi di dimensioni maggiori) ed un moderato ingrossamento della milza. Quando ereditata in combinazione con la ß-talassemia, può portare a forme più gravi di malattia.
Emoglobina H (HbH) e Emoglobina di Bart
Queste emoglobine anomale si formano specificamente in alcuni casi di alfa-talassemia, a causa della grave carenza di catene alfa.
- Emoglobina H (HbH): È composta da quattro catene beta globiniche (β4). Si forma in individui con alterazione di tre dei quattro geni alfa-globinici.
- Emoglobina di Bart: È composta da quattro catene globiniche gamma (γ4). Si sviluppa nei feti affetti da alfa-talassemia grave, in cui tutti e quattro i geni alfa-globinici sono alterati, causando una condizione spesso letale.
Diagnosi delle Emoglobinopatie: Un Approccio Multifattoriale
La diagnosi delle emoglobinopatie è un processo complesso che richiede una valutazione complessiva di una serie di esami, eseguiti secondo processi ben definiti, poiché l'esecuzione di un singolo test non è sufficiente. È fondamentale prestare molta attenzione all'interpretazione dei risultati, data la variabilità e la complessità di queste patologie.
Test di Primo Livello: L'Assetto Emoglobinico
I test di primo livello per la ricerca delle emoglobinopatie in genere utilizzano metodiche volte alla determinazione del tipo e della quantità di emoglobine presenti nel sangue del paziente in esame. Questo esame, noto come "assetto emoglobinico" o "elettroforesi dell'emoglobina", si prefigge di rilevare eventuali varianti emoglobiniche e/o la quantità relativa dei diversi tipi di Hb (HbA, HbA2, HbF, e altre varianti).
La maggior parte delle varianti emoglobiniche più comuni o delle talassemie possono essere identificate utilizzando una combinazione di questi test. La rilevazione della quantità relativa della variante emoglobinica presente è un valido ausilio diagnostico, fornendo indicazioni importanti sulla presenza e sulla gravità potenziale di una emoglobinopatia. I risultati in genere riportano il tipo di Hb presenti (quando identificabile) e la loro quantità relativa.
Test Molecolari
Per una diagnosi più precisa e per identificare mutazioni specifiche, vengono impiegati i test molecolari. Questi test ricercano direttamente le mutazioni presenti nei geni codificanti per le catene globiniche alfa e beta. I test molecolari sono particolarmente utili per confermare una diagnosi sospetta, per la consulenza genetica e per la diagnosi prenatale, specialmente in casi complessi o in presenza di eterozigosi composta.
Screening Neonatale
Il test per la ricerca delle emoglobinopatie viene richiesto come parte dei programmi di screening neonatale, attualmente attivo solo in alcune regioni in Italia. Lo screening delle emoglobinopatie consente di identificare e quindi potenzialmente anche trattare i neonati con disordini congeniti entro pochi giorni dalla nascita. In questo modo possono essere evitati problemi di salute potenzialmente letali o anche responsabili di disabilità permanenti, migliorando significativamente la prognosi a lungo termine per i bambini affetti. L'esame può essere richiesto anche nel caso in cui si sospetti un'emoglobinopatia in presenza di segni e sintomi caratteristici, indipendentemente dall'età del paziente.
Considerazioni Diagnostiche Importanti
È cruciale considerare alcuni fattori che possono interferire con la valutazione delle emoglobinopatie. Le trasfusioni di sangue, ad esempio, possono alterare i risultati, poiché il metodo analitico non è in grado di distinguere tra l'emoglobina del donatore e quella del paziente, interferendo potenzialmente con i risultati del test. Per ottenere risultati accurati, un paziente dovrebbe aspettare diversi mesi dopo una trasfusione prima di sottoporsi a questo esame. È anche importante notare che l'esame per l'emoglobina fetale e l'assetto emoglobinico non vengono eseguiti in tutti i laboratori, e può essere necessario rivolgersi a centri specializzati.
Significato Clinico dei Livelli di Emoglobina Fetale: Quando l'HbF Ritorna Protagonista
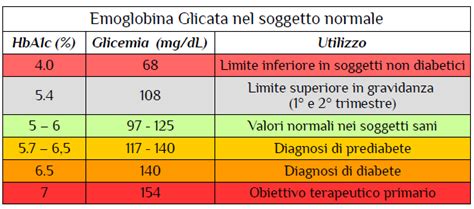
L'emoglobina fetale (HbF), pur essendo predominante nella vita fetale e declinando rapidamente dopo la nascita, mantiene un ruolo significativo anche nell'età adulta, soprattutto in contesti patologici. Una piccola percentuale di emoglobina fetale viene espressa anche durante la vita adulta ed i suoi livelli possono variare anche di molto sotto l'influenza di fattori quali l'età, il sesso o peculiarità genomiche.
In un adulto, livelli di emoglobina fetale considerabili normali oscillano tra 0.1 e 1.1% (o tra 0.3% e 1.2% a seconda della metodica analitica). Quando negli adulti si hanno valori superiori a 1.1%, si parla di emoglobina F alta. Un'anomalia nei livelli delle frazioni dell'emoglobina può indicare la presenza di emoglobinopatie. Il dosaggio delle frazioni dell'emoglobina rappresenta uno strumento potente per la diagnosi, il monitoraggio e la gestione delle emoglobinopatie.
Elevate proporzioni di emoglobina F sono presenti in diverse condizioni patologiche, tra cui le talassemie e altre anemie croniche. L'aumento dell'HbF in questi contesti può rappresentare un meccanismo compensatorio del corpo per migliorare il trasporto di ossigeno quando la produzione di emoglobina adulta è compromessa. Una condizione in cui l'emoglobina F può essere alta, ma è necessaria una precisazione, è una condizione di recupero dovuta a ipoplasia di midollo osseo, ovvero un disturbo delle cellule staminali ematopoietiche.
Persistenza Ereditaria dell'Emoglobina Fetale (HPFH)
Alcuni soggetti sono affetti dalla cosiddetta persistenza ereditaria dell'emoglobina fetale (HPFH), una condizione benigna in cui concentrazioni importanti di emoglobina fetale (superiori al 10%) persistono anche in età adulta. Questa peculiarità, generalmente asintomatica, può avere un impatto sorprendente e positivo, in quanto si è notato come possa alleviare la severità di certe emoglobinopatie e talassemie.
La HPFH associata a beta talassemia è caratterizzata da livelli elevati di emoglobina fetale (HbF) e da un aumento del numero delle cellule contenenti l'HbF. La prevalenza di questa forma non è nota. L'associazione tra la HPFH e la ß-talassemia attenua i sintomi clinici nei pazienti, che possono essere asintomatici o presentare una ß-talassemia intermedia, riducendo la necessità di trasfusioni e migliorando la qualità di vita. La HPFH è dovuta a delezioni nel cluster genico della beta-globina o a mutazioni puntiformi nei geni HBG1 e HBG2 (sul cromosoma 11p15.5), che regolano l'espressione delle catene gamma.
La diagnosi di HPFH si basa sulla presenza di un significativo aumento dell'HbF, che varia tra il 10% e il 40% negli eterozigoti, con indici eritrocitari normali o quasi. L'HbF si distribuisce in maniera omogenea negli eritrociti e i livelli dell'HbA2 sono normali o ridotti. La trasmissione della HPFH è co-dominante. La differenziazione tra la HPFH e la delta-beta talassemia può essere difficile e può richiedere il rapporto di sintesi tra le globine alfa-beta e l'esame del DNA; non sempre questa differenziazione è possibile sulla base dell'analisi standard del sangue.
Terapie e Gestione delle Emoglobinopatie: Il Ruolo Terapeutico dell'HbF
Il trattamento di alcuni tipi di emoglobinopatie può comportare l'utilizzo di terapie di supporto, ad esempio durante la comparsa di crisi in corso di anemia falciforme. L'obiettivo primario di queste terapie è quello di alleviare il dolore e di minimizzare le complicanze acute e croniche della malattia. Talvolta, in caso di anemia grave, è necessario ricorrere a trasfusioni di sangue per mantenere livelli adeguati di emoglobina e prevenire l'ipossia tissutale. Meno frequentemente, possono essere utilizzati anche altri trattamenti, a seconda della specifica emoglobinopatia e della sua gravità.
Una delle strategie terapeutiche più innovative e promettenti è quella di aumentare la concentrazione di emoglobina fetale (HbF) nei pazienti affetti da determinate emoglobinopatie. Una terapia farmacologica capace di aumentare la concentrazione di emoglobina fetale apporta benefici significativi ad alcune categorie di pazienti, come quelli affetti da anemia falciforme o da talassemia Beta, sfruttando la capacità dell'HbF di compensare la disfunzione dell'emoglobina adulta patologica.
Il prototipo di questi farmaci è stata l'idrossiurea, un farmaco antineoplastico ad azione mielosoppressiva, che si è dimostrato efficace nell'aumentare i livelli di emoglobina fetale. Questo aumento dell'HbF, specialmente nei pazienti con anemia falciforme, porta a una riduzione dell'incidenza di crisi dolorose, poiché l'HbF interferisce con la polimerizzazione dell'HbS, prevenendo la falcizzazione dei globuli rossi. Analogamente, nei pazienti con ß-talassemia, l'incremento di HbF può ridurre la necessità di trasfusioni di sangue, alleviando la gravità della malattia.
La ricerca biomedica continua a esplorare nuove vie per modulare l'espressione dell'HbF, con l'obiettivo di sviluppare terapie ancora più mirate ed efficaci. Ad esempio, gli studi si concentrano sull'inibizione di BCL11A, un fattore di trascrizione che agisce come repressore principale della produzione di HbF nell'adulto. La modulazione di BCL11A rappresenta una strategia promettente per riattivare la sintesi di emoglobina fetale, offrendo nuove prospettive per il trattamento di queste malattie genetiche del sangue.
Come si Effettua l'Esame dell'Emoglobina Fetale e Preparazione
L'esame per l'emoglobina fetale, come parte dell'assetto emoglobinico, viene svolto per la diagnosi di patologie a carico del sangue, quali talassemia e anemia falciforme, e per monitorare i livelli di HbF in adulti e bambini in contesti clinici specifici.
Per l'esame dell'emoglobina fetale, viene richiesto un prelievo di sangue venoso. Non è una procedura complessa e si svolge come un normale prelievo ematico. Tuttavia, per garantire l'accuratezza dei risultati, è generalmente richiesto un digiuno di almeno otto ore prima del prelievo. È sempre consigliabile informare il medico o il laboratorio su eventuali farmaci assunti, trattamenti in corso o recenti trasfusioni di sangue, poiché questi fattori possono influenzare l'interpretazione dei risultati. I valori di riferimento degli esami di laboratorio possono variare a seconda della metodologia di analisi dei campioni utilizzata dal laboratorio; quelli indicati in questa scheda hanno uno scopo puramente informativo e devono sempre essere interpretati da un professionista sanitario nel contesto clinico del paziente.
tags: #emoglobina #fetale #agora #scienze #biomediche