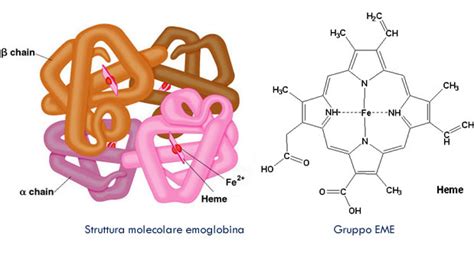
L'emoglobina è una proteina essenziale che si trova nei globuli rossi, deputata al trasporto dell'ossigeno dai polmoni ai tessuti dell'organismo. Ogni molecola di emoglobina è un complesso macchinario molecolare, composto da quattro subunità proteiche globulari, note come catene, a cui è strettamente associata una molecola non proteica chiamata eme. L'eme, un anello tetrapirrolico contenente un atomo di ferro, è il sito attivo dove l'ossigeno si lega.
L'Emoglobina nell'Adulto e nel Feto: Strutture Distinte
Nell'adulto, la forma predominante di emoglobina è l'HbA, costituita da due catene alfa (α) e due catene beta (ß). Tuttavia, esistono anche altre forme in percentuali minori, come l'HbA2 (composta da due catene α e due catene delta, δ) e l'HbF, l'emoglobina fetale. Quest'ultima è la forma dominante durante la vita fetale ed è caratterizzata dalla presenza di due catene alfa (α) e due catene gamma (γ). Dopo la nascita, i livelli di HbF diminuiscono progressivamente, attestandosi sulle percentuali tipiche dell'adulto entro i primi sei mesi di vita.
La Struttura dell'Emoglobina Fetale (HbF)
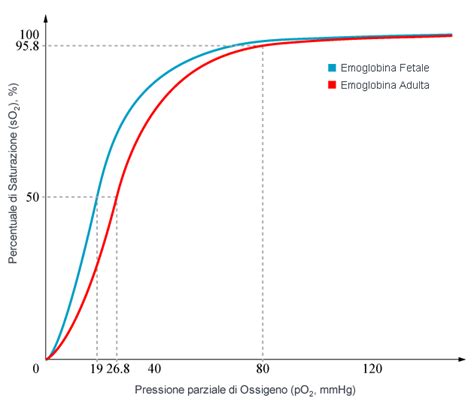
L'emoglobina fetale, o HbF, presenta una struttura molecolare leggermente diversa da quella adulta. È formata da due catene α, identiche a quelle dell'emoglobina adulta, e da due catene γ. Ciascuna catena γ è composta da 146 amminoacidi, mentre le catene β dell'emoglobina adulta ne contengono 141. La differenza risiede in 39 amminoacidi specifici che compongono le catene γ rispetto alle catene β. Questa modifica strutturale conferisce all'HbF un'affinità per l'ossigeno superiore rispetto all'emoglobina adulta. In altre parole, l'HbF si lega all'ossigeno in modo più tenace.
Il Ruolo Funzionale dell'HbF nella Vita Fetale
Dal punto di vista funzionale, la maggiore affinità dell'HbF per l'ossigeno è cruciale per la sopravvivenza fetale. Permette al feto di estrarre con maggiore efficacia l'ossigeno dal sangue materno attraverso la barriera placentare. Questo trasferimento di ossigeno è ulteriormente favorito da una concentrazione di emoglobina nel sangue fetale circa il 50% più alta rispetto a quella del sangue materno.
Sviluppo delle Catene Globiniche: Un Processo Geneticamente Regolato
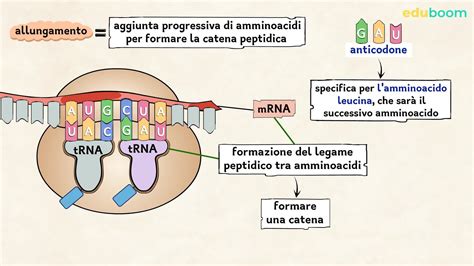
L'espressione delle diverse catene globiniche umane, che compongono le varie forme di emoglobina, è un processo finemente regolato a livello genetico. L'informazione genetica per la sintesi di queste catene è codificata nel DNA e viene trascritta in mRNA, che a sua volta dirige la sintesi delle catene polipeptidiche composte da specifici amminoacidi.
La sintesi delle catene globiniche β, che caratterizzano l'emoglobina adulta, è appena percettibile durante la vita fetale. Raggiunge il suo regime normale solo verso la fine del terzo mese di vita extrauterina. Le concentrazioni di emoglobina fetale, invece, diminuiscono drasticamente entro il primo anno di vita, scendendo a livelli generalmente inferiori all'1%. Negli adulti normali, i valori di emoglobina fetale sono compresi tra lo 0.3% e l'1.2%, con meno del 3.5% di emoglobina A2 e la restante percentuale (>96%) costituita da emoglobina di tipo A.
La differente espressione temporale delle catene globiniche, dal concepimento alla vita adulta, è determinata dall'attivazione e dallo spegnimento di specifici geni.
Emoglobinopatie e Talassemie: Alterazioni Genetiche con Conseguenze Cliniche
Le alterazioni nella sintesi delle catene globiniche possono portare a una serie di disturbi noti come emoglobinopatie e talassemie.
Le talassemie sono causate da difetti ereditari che compromettono la normale sintesi delle catene α (α-talassemia) o delle catene ß (ß-talassemia).
α-talassemia: L'informazione genetica per le catene α è contenuta in quattro geni. Un difetto in tutti e quattro i geni è incompatibile con lo sviluppo fetale. L'alterazione di tre geni porta alla presenza di emoglobina H (HbH), formata da complessi di quattro catene ß, accompagnata da anemia emolitica e splenomegalia. La delezione di due geni causa il tratto talassemico, con anemia, globuli rossi di volume ridotto e livelli normali di HbA2 e HbF. L'alterazione di un solo gene è spesso indistinguibile dalla normalità, ma può comportare la trasmissione del carattere alla prole.
ß-talassemia: L'informazione genetica per le catene ß è contenuta in due geni. L'alterazione di entrambi i geni (forma omozigote) porta alla ß-talassemia major, o morbo di Cooley. Questa grave forma di anemia si manifesta subito dopo la nascita e richiede trasfusioni di sangue periodiche. L'alterazione di un solo gene (forma eterozigote) è definita ß-talassemia minor o tratto talassemico. La maggior parte degli individui con talassemia minor non presenta sintomi evidenti; i loro globuli rossi sono più numerosi ma di volume ridotto (microcitemia) e poveri di emoglobina.
Un esempio di emoglobinopatia è l'anemia falciforme, causata da una mutazione genetica che porta alla sostituzione della valina con l'acido glutammico nella posizione 6 della catena ß. Questo difetto porta alla formazione di emoglobina anomala (HbS) che, in condizioni di bassa tensione di ossigeno, fa sì che i globuli rossi assumano una forma a falce, portando a occlusioni vascolari e dolore.
Terapia Genica per la Beta Talassemia: pazienti liberi dalle trasfusioni per sempre
Significato Patologico e Potenziali Terapie
Nell'utero, un feto normale produce una piccola quota di emoglobina adulta (2.5-5%). Un feto affetto da talassemia major ne produce una quantità ancora inferiore (<2%). La determinazione della quantità di emoglobina adulta in un campione di sangue fetale, prelevato tramite cordocentesi, può essere utilizzata per diagnosticare la talassemia major durante la gravidanza.
Una piccola percentuale di emoglobina fetale viene espressa anche in età adulta, e i suoi livelli possono variare in base a fattori come età, sesso e peculiarità genomiche. Alcuni individui presentano la persistenza ereditaria dell'emoglobina fetale, una condizione benigna in cui concentrazioni significative di HbF (>10%) persistono in età adulta. Questa peculiarità, generalmente asintomatica, può mitigare la gravità di alcune emoglobinopatie e talassemie.
Terapie farmacologiche volte ad aumentare la concentrazione di emoglobina fetale hanno dimostrato benefici significativi in pazienti affetti da anemia falciforme e ß-talassemia. L'idrossiurea, un farmaco antineoplastico con azione mielosoppressiva, è stata utilizzata con successo per incrementare i livelli di HbF e ridurre l'incidenza di crisi dolorose nei pazienti con anemia falciforme.
Implicazioni Molecolari e Funzionali
La comprensione della relazione tra geni ed effetti a livello molecolare è fondamentale per chiarire le basi delle malattie del sangue. La sintesi proteica segue il dogma centrale della biologia molecolare: DNA → mRNA → catena polipeptidica. Le catene polipeptidiche, composte da specifici amminoacidi, si ripiegano in strutture tridimensionali complesse per formare proteine funzionali. La sequenza amminoacidica determina la struttura e, di conseguenza, la funzione della proteina. Un'alterazione nella sequenza amminoacidica, anche minima, può avere effetti di vasta portata sullo sviluppo o sulle funzioni dell'organismo, come nel caso dell'anemia falciforme, dove una singola sostituzione amminoacidica porta alla formazione di emoglobina anomala con gravi conseguenze cliniche. Le proteine, inclusa l'emoglobina, possono anche far parte di altre molecole complesse, ampliando ulteriormente la loro importanza biologica. L'emoglobina umana, ad esempio, ha un peso molecolare di circa 64.000 Dalton e le sue molecole sono di dimensioni di circa 4,4 x 4,4 x 2,5 nm, rendendole facilmente distinguibili alla semplice osservazione al microscopio elettronico. La comprensione di questi processi molecolari è cruciale per lo sviluppo di strategie diagnostiche e terapeutiche per le malattie genetiche.
tags: #curva #saturazione #hb #fetale